|
Leukaemia Research Fund Centre, Institute of Cancer Research, Chester
Beatty Laboratories,
London SW3 6JB Mel Greaves professor of cell biology
m.gr.eaves@icr.acuk
BMJ 2002;324:283-7
Molecular genetics provide
exciting new insights into the pathogenesis of childhood leukaemia
The risk of any child developing acute leukaemia is about 1 in 2000
with 400-450 new cases a year in the United Kingdom. Cure rates
approaching 75% can be achieved with combination chemotherapy, but
this figure disguises success rates that vary from 10% to 90% with
the different biological subtypes of the disease. In this review
I discuss how new insights into the underlying molecular biology
of leukaemia have changed our understanding of the disease. Not
only is there the prospect of better treatment and the introduction
of new biologically based therapies, but, as the causes of disease
are being unravelled, the possibility of prevention may not just
be wishful thinking.
Methods
This article is based on information and views published from my
laboratory plus comprehensive, prospective screening of leading
journals for leukaemia and cancer research.
A diverse disease with variable clinical outcome
It has long been recognised that childhood leukaemia is not one
homogeneous disease. The major morphological division into acute
lymphoblastic leukaemia and acute myeloblastic leukaemia is supplemented
by the identification of a range of subsets based on gene expression,
antigens that delineate cell type or differentiation status, and
chromosomal and molecular abnormalities. These include chromosome
translocationsexchanges of large tracks of DNA between chromosomes,
resulting (at the point of exchange) in the generation of chimeric
or fusion genes1-and changes in chromosome number (hyperdiploidy
or hypodiploidy). At a more subtle level, there may also be gene
deletions or single nucleotide base changes in genes.
This molecular archaeology has uncovered what has long been suspected-that
the acute leukaemias are biologically diverse diseases. ² Moreover,
in acute lymphoblastic leukaemia these subgroups segregate with
age (fig I), which may help explain the considerable differences
in outcome between infants aged < 1 year, children (2 -10 years
old), and adults. A similar spectrum of molecular diversity exists
for acute myeloblastic leukaemia. Several of these molecular abnormalities
have independent prognostic importance in the context of particular
treatment regimens (see table). ²
Summary points
Different chromosomal and gene abnormalities in leukaemia define
biological subsets of disease
with prognostic importance
Chromosome translocations generate chimeric fusion genes, which
provide stable sensitive
markers that are unique for each patient's leukaemic clone and can
be used to track its
origins and Tesponse to treatment
The common chromosome translocations in childhood leukaemia
seem to initiate disease
and often arise prenatally
One or more postnatal genetic alterations are also needed for
leukaemia development;
and in childhood acute lymphoblastic leukaemia these may be caused
by abnormal
immune responses to infection
Proteins coded by fusion genes operate principally by blocking
cell differentiation
in leukaemic cells and provide potential targets for new treatments
What most descriptions of leukaemic cells obscure, however, is
the dynamic, evolving nature of the disease, a feature it shares
with all other cancers: Leukaemia is a clonal disease (originating
in a single cell) and evolves by the accrual of mutations within
a clone. This results in progressive genetic diversification followed
by a "natural selection" of dominant mutant subclones.
Clinical outcome depends on not only the nature of the leukaemic
clone but how far it has evolved by the time pathological symptoms
are recognised, a correct diagnosis is made, and treatment started.
Diagnostic delay increases the probability that the clone will have
progressed to the point where additional mutations have been acquired,
including those endowing drug resistance, rendering eradication
more difficult.
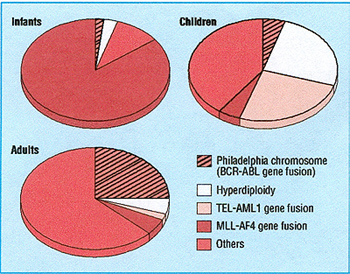
Fig 1 Major molecular subsets of acute lymphoblastic leukaemia
in infants (1 year old), children (2-10 years old), and adults

The fetal origins of childhood leukaemia
There is now compelling evidence that chromosome translocations
are often the first or initiating events inleukaemia, occurring
prenatally during fetal development This evidence comes from two
sources-identical twin infants or children with concordant acute
lymphoblastic leukaemia (5 6 ) and retrospective scrutiny of neonatal
blood spots or Guthrie cards: H
The most common structural genetic abnormality in childhood leukaemia
is a fusion of two genes, TEL and AML1, This is generated by a chromosome
translocation between cromosomes 12 and 21. Simultaneous breaks
in the TEL gene {chromosome 12) and AML1 gene {chromosome 21) are
followed by error-prone repair that stitches up the DNA across chromosomes
12 and 21, joining the normally separate TEL and AML 1 genes together
to form a chimeric or fusion gene {fig 2). As in other chromosomal
translocations, the DNA breaks always occur in non-coding regions
{introns) of genes. The precise breakpoints in the TEL and AML 1
genes can be identified or mapped by the "long distance"
polymerase chain reaction {PCR). Breaks always occur, more or less
randomly, within a limited region of these genes, but each patient's
leukaemic cells have a unique {or clone specific) breakpoint in
the DNA sequence.
Analysis of pairs of identical twins with concordant acute lymphoblastic
leukaemia shows that leukaemic cells from both twins in a pair share
the identical breakpoints in TEL and AML 1 genes or, in the case
of
infant twins with acute lymphoblastic leukaemia, the same breakpoints
in the MLL gene.(5 6) Monozygotic twins are, of course, themselves
monoclonal and genetically identical, but gene breakpoints in leukaemic
cells are not inherited-they disappear in
remission.
The only plausible explanation for twin leukaemias
sharing the same gene breakpoints is that the d1romosomal breaks
generating the fusion gene must have occurredjust once, in one blood
stem cell, in one twin in utero. Subsequently, but still in utero,
descendent progeny of this transformed cell spread to the other
twin, presumably via the anastomoses that exist within shared, single
{monod1orionic) placentas.5 We assume that at this early stage a
clinically silent or covert preleukaemic clone is generated which,
after birth, may evolve to full blown leukaemia anything from two
(3 6) months to 14 years later:
Further evidence that childhood leukaemia Call
originate before birth comes from scrutiny of neonatal blood spots
or Guthrie cards {fig 3). PCR tests for specific fusion genes, designed
for each patient, can detect as few as 1-20 leukaemic cells in a
blood spot. The presence of the same fusion gene sequence in a neonatal
blood spot as is in the patient's leukaemic cells at diagnosis 7
8 provides unequivocal evidence that

Fig 2 Chromosomal translocation to form the TEL -AML
1 fusion gene in childhood acute lymphoblastic leukaemia. Top: Fluorescence
in situ hybridisation labelling of dividing leukaemic cell chromosomes
with specific probes for chromosome 12 (red) and chromosome 21 (green)
reveal two red and green chromosomes (one large, one small). These
are copies of chromosomes 12 and 21 between which there has been
a reciprocal exchange of DNA. Bottom: The TEL and AML 1 genes lie
at the breaks and are brought together by the exchange. The genes
break in non-coding (grey) regions between the coding regions (numbered,
green or red), and re-joining of the two broken genes forms a novel
fusion gene

Fig 3 Identification of fusion genes in neonatal blood
spots of patients with leukaemia. At diagnosis of childhood acute
lymphoblastic leukaemia, a TEL -AML 1 fusion gene can be identified
in the leukaemic cells. The TEL -AML 1 sequence is first determined
by long range PCR, then oligonucleotide primers are designed for
that unique sequence and for use in short range (conventional) PCR.
DNA is extracted from a diagnostic sample for PCR and, in parallel,
a segment from a neonatal blood spot is subjected to PCR. If successful,
both samples from the patient amplify to produce a nucleotide sequence
visualised as a band in a gel. Sequencing of these bands shows them
to be identical
leukaemia has been initiated prenatally, probably by formation
of the fusion gene itself. The conclusions from Guthrie card studies
is that leukaemia is fetal in origin in all cases of infant leukaemia
( with fusions of the MLL gene), in most cases of the common form
of childhood acute lymphoblastic leukaemia (with TELAML 1 ), and
in about half of cases of childhood acute myeloblastic leukaemia
(with translocation of cromosomes 8 and 21).
The "two hit" model for childhood leukaemia
Although there are no accurate data for concordance rates of leukaemia
in infant twins, anecdotally it seems to be exceptionally hiR"h,
perhaps approad1inR" 100% (that is, if one twin has it, so
will the other). If correct, this suggests that MLL gene fusion
in utero has a dramatic impact, ensuring subsequent leukaemia. But
for children aged 2-6 years (with acute lymphoblastic leukaemia)
the concordance rate is considerably lower, around 5%0. This still
represents a lOO-fold extra risk of leukaemia for the twin of a
patient with acute lymphoblastic leukaemia but also indicates the
need for some additional postnatal event(s) for which there is a
1 in 20 chance, or 95% discordance. This suggests, at a
minimum, a "two hit" model for the natural course of childhood
leukaemia (fig 4).3
If this model of leukaemia development is correct, then, for every
child with acute lymphoblastic leukaemia diagnosed, there should
be at least 20 healthy children who have had a chromosome translocation,
a functional leukaemia fusion gene, and a covert preleukaemic clone
generated in utero. This possibility has been investigated by screening
unselected samples of newborn cord blood for fusion genes. About
600 samples have been screened, and around 1 %0 have a leukaemic
TEL-AMLl fusion gene (H Mori, et al, personal communication). This
1% represents 100 times the cumulative rate or risk of acute lymphoblastic
leukaemia (with a TEL-AMLl gene), indicating that the frequency
of conversion of the preleukaemic clone to overt disease is low.
The real bottleneck in development of acute lymphoblastic leukaemia
therefore seems to be a stringent requirement for a second "hit"
after birth-that is, exposure and additional chromosomal or molecular
abnormality.
Causal mechanisms
A key issue to resolve is what exposures or events might precipitate
the cromosome breaks whose improper repair initiates or promotes
childhood leukaemia. Given the biological diversity of leukaemia,
it is highly unlikely that there is a single cause. Even for a defined
biological subtype of the disease, there probably isn't one cause
as such but a causal mechanism. As with other cancers, this is likely
to involve an interaction of exposure (exogenous or endogenous)
with inherent genetic susceptibility, and d1ance4
Epidemiological evidence suggests that ionising radiation, certain
chemicals (such as benzene), viruses (human T cell leukaemia/lymphoma
virus type I, Epstein-Barr virus), and bacteria (Helicobacter pylori)
may playa part in the development of some subtypes of leukaemia
and lymphoma in adults and children. Whether any of these exposures
have a major role in childhood leukaemia is uncertain, but large
scale casecontrol molecular epidemiological studies in Britain and
the United States may provide answers. The UK children's cancer
study (UKCCS) seeks to address several hypotheses on different exposures,
combined with definition of biological subtypes of disease and genetic
studies. 9 It and a parallel US study have already ruled out electromagnetic
fields as a major factor in leukaemia aetiology.10
A critical role for infection?
Two hypotheses have suggested that an abnormal response to common
infections plays a decisive role in the development of childhood
acute lymphoblastic

Fig 4 Natural course of childhood leukaemia
leukaemia. One proposes that transiently increased rates of leukaemia
(of any subtype), sometimes in clear geographical clusters, are
due to population mobility and mixing resulting in infection occurring
in previously unexposed or susceptible individuals.11 The "delayed
infection" hypothesis suggests that acute lymphoblastic leukaemia
in children is caused by a lack of exposure to infection and a failure
of immune system modulation in infancy.12 Later, an abnormal immune
response occurs to one or more common bacterial or viral infections
incurred after ( delayed) mixing with infectious carriers, such
as other children in playgroups or schools.
This second hypothesis is similar to the "hygiene hypothesis"
put forward to explain allergies and asthma and type I diabetes.13
It suggests that it is the aberrant response to infection that promotes
the crucial second, postnatal event 12 Epidemiological sup
port for the delayed infection hypothesis come from studies of children
with acute lymphoblastic leukaemia. These show that such children
are less likely to have had some common infections in infancy, have
had fewer social contacts in infancy, are more likely to have been
first born, and are less likely to have received certain vaccinations,
particularly for Haemophilus influenzae.14 15
Over the next year or two, it should become clear whether childhood
leukaemia involves infectious exposures. If it does, this raises
the possibility of prevention, but it cannot apply to all types
of childhood leukaemia. For acute lymphoblastic leukaemia in infants
(with MLL gene fusions), aetiological hypotheses and the available
epidemiological data are distinct A recent international epidemiological
study of infant leukaemia has implicated transplacental chemical
exposures to pesticides (Baygon) and a drug ( dipyrone) during pregnancy.16
Inherited susceptibility
Inherited genetic variation is likely to be important in determining
differential susceptibility to leukaemia, as in other cancers and
diseases. Risk of infant leukaemia has been associated with polymorphic
variants of the NQO I gene, which codes for an enzyme that detoxifies
benzene metabolites and quinone-containing flavonoids and other
substances.17 For typical childhood acute lymphoblastic leukaemia,
there is preliminary evidence that HLA class II alleles influence
risk,18 and inherited variations in other immune genes that influence
responses to infection probably playa role in susceptibility. Finally,
risk of acute lymphoblastic leukaemia in infants, older children,
and adults has been linked to inheritance of alleles of MTHFR, a
key gene in the folate metabolism pathway.19 The reason for this
association may lie in the way that folate metabolism affects the
fidelity of DNA replication and, possibly, vulnerability to chromosomal
breaks. Recent data suggest that folate intake in pregnant women
may be an important dietary modifier of risk for paediatric acute
lymphoblastic leukaemia.20
Additional education resources
.Leukaemia Research Fund (www.lrf.org.uk).
Information on booklets available on leukaemia for parents and
patients as well as both
reference works and books for medical students and doctors
.Seminars in Haematology.2000;37(4). Review articles on chromosome
changes in leukaemia and related diseases
.Henderson ES, Lister TA, Greaves MF,eds. Leukemia. 6th ed. Philadelphia:
WB Saunders,1996.Standard textbook on leukaemia covering biology
and
treatment in adults and children
New therapeutic targets and better indicators of prognosis
Genetic alterations in leukaemia (and other cancers) affect the
complex signalling networks that control cellproliferation, cell
differentiation, and cell death by apoptosis.21 These effects help
to explain why certain molecular abnormalities result in adverse
clinical outcomes. They also present new opportunities for targeting
treatments. For example, the BCR-ABL fusion gene (found in the Philadelphia
chromosome associated with acute lymphoblastic leukaemia and chronic
myeloid leukaemia) results in the production of an active kinase
enzyme (from the ABL part of the gene) that drives cell proliferation
independently of normal requirements for growth factor and blocks
apoptosis and therefore drug responsiveness pathways. Normal p53
protein in cells is required to induce cell death after anoxia or
DNA damage from exposure to drugs or irradiation. Mutations or deletions
in the p53 gene are rare at presentation of leukaemia but are more
common at relapse, helping to explain the therapeutic "resistance"
of more advanced disease.
The fusion genes generated by chromosome translocation (TEL-AML
I in acute lymphoblastic leukaemia, AMLI-ETO in acute myeloblastic
leukaemia, and PML-RARA in acute promyelocytic leukaemia) primarily
block cell differentiation. The aberrant proteins produced by these
genes inhibit gene activity and differentiation by recruiting repressor
molecules.22 These repressors include histone deacetylase enzymes.
These enzymes can, however, be counteracted by selective inhibitors,
and one promising line of treatment-at present targeted at acute
promyeloblastic leukaemia
with PML- RARA fusions-is to use such drugs to reverse the block
to normal cell development 23 The
success in using a derivative of retinoic acid to induce remission
in acute promyeloblastic leukaemia24 is also encouraging in this
respect
Another promising approach is the use ofSTI-571, a selective inhibitor
of Abl protein and related kinases,
which is has been shown to have a major impact on chronic myeloid
leukaemia.25 26 Hopefully, other small
compounds can be designed that will block the signalling pathways
that are overactive in paediatric and adult leukaemic cells, preferably
in a non-toxic fashion as with STI-571.
Another hope is that the ability to obtain more comprehensive or
complete genetic profiles of leukaemic cells may allow prognosis
to be defined extremely accurately.27 Prognosis in paediatric acute
lymphoblastic leukaemia can already be assessed with some accuracy
using highly sensitive polymerase chain reaction methods to detect
molecular markers of leukaemic cells, such as clonal rearrangements
of immunoglobulin genes or T cell receptor genes or the unique leukaemia
fusion genes. With such methods, quantitative assessment of levels
of residual leukaemic cells during treatment is predictive of later
outcome28 and may therefore provide a good guide to individual patient
management
I thank Dr C Harrison for supplying the figure of fluorescence
in situ hybridisation labelling of leukaemic cell chromosomes.
Funding: Work in my laboratory is supported by the Leukaemia Research
Fund and the Kay Kendall Leukaemia Fund.
Competing interests: None declared.
1 Rowley JD. The critical role of chromosome translocations in
human
leukemias. Annu Rev Genet I 998;32:495-51 9.
2 Kersey JH. Fifty years of studies of the biology and therapy of
childhood
leukemia. Blood1997;90:4243-51.
3 Greaves M. Molecular genetics, natural history and the demise
of
childhood leukaemia. EUT J Cancer 1999;35: 173-85.
4 Greaves M. Cancer. The evolutionary legacy. Oxford: Oxford University
Press, 2000.
5 Ford AM, Ridge SA, Cabrera ME, Mahmoud H, Steel CM, Chan LC, et
al.
In utero rearrangements in the trithorax-related oncogene in infant
leukaemias. Nature 1993;363:358-60.
6 Wiemels JL, Ford AM, Van Wering ER, Postma A, Greaves M. Protracted
and variable latency of acute lymphoblastic leukemia after TEL-AML1
gene fusion in utero. Blood1999;94:1057-62.
7 Gale KB, Ford AM, Repp R, Borkhardt A, Keller C, Eden OB, et al.
Backtracking leukemia to birth: identification of clonotypic gene
fusion sequences in neonatal blood spots. Proc Natl Acad Sci USA
1997;94:13950-4.
8 WiemelsJL, Cazzaniga G, Daniotti M, Eden OB, Addison GM, Masera
G, et al. Prenatal origin of acute lymphoblastic leukaemia in children.
Lancet 1999;354: 1499-503.
9 UK Childhood Cancer Study Investigators. The United Kingdom childhood
cancer study: objectives, materials and methods. BT J Cancer 2000;82:
1073-102.
10 UK Childhood Cancer Study Investigators. Childhood cancer and
residential proximity to power lines. BT J Cancer 2000;83:1573-80.
11Kinlen LJ. Epidemiological evidence for an infective basis in
childhood
leukaemia. BT J Cancer 1995;71:1-5.
12 Greaves MF. Aetiology of acute leukaemia. Lancet 1997 ;349:344-9.
13 Wills-Karp M, Santeliz J, Karp CL. The germless theory of allergic
disease: revisiting the hygiene hypothesis. Nat Rev 200 1;1 :69-
75.
14 Dockerty JD, Draper G, Vincent T, Rowan SD, Bunch KJ. Case-control
study of parental age, parity and socioeconomic level in relation
to childhood cancers. Int J Epidemiol (in press ).
15 Auvinen A, Hakulinen T Groves F. Haemophilus influenzae type
B vaccination and risk of childhood leukaemia in a vaccine trial
in Finland. BT J Cancer 2000;83:956-8.
16 Alexander FE, Patheal SL, Biondi A, Brandalise S, Cabrera M-E,
Chan LC, et al. Transplacental chemical exposure and risk of infant
leukemia with MLL gene fusion. CancerRes 2001;61:2542-6.
17 Wiemels JL, Pagnamenta A, Taylor GM, Eden OB, Alexander FE, Greaves
MF, et al. A lack of afunctional NAD(P)H:quinone oxidoreductase
allele is selectively associated with pediatric leukemias that have
MLL fusions. Cancer Res 1999;59:4095-9.
18 Taylor GM, Dearden S, Payne N, Ayres M, Gokhale DA, BirchJM,
et al. Evidence that an HLA-DQAI-DQB1 haplotype influences susceptibility
to childhood common acute lymphoblastic leukaemia in males provides
further support for an infection-related aetiology. BT J Cancer
1998;78:561-5.
19 Wiemels JL, Smith RN, Taylor GM, Eden OB, Alexander FE, Greaves
MF, et al. Methylenetetrahydrofolate reductase (MfHFR) polymorphisms
and risk of molecularly defined subtypes of childhood acute leukemia.
ProcNatlAcadSci USA 2001;98:4004-9.
20 ThompsonJR, FitzGerald P, Willoughby MLN, Armstrong BK. Maternal
folate supplementation in pregnancy and protection against common
acute lymphoblastic leukaemia in childhood: a case-control study.
Lancet 200 1;358: 1935-40.
21 Hanahan D, Weinberg RA.The hallmarks of cancer. Cell2000;100:57-70.
22 Guidez F, Zelent A Role of nuclear receptor co-repressors in
leukemogenesis. CurT ToP Microbiol Immunol200 1;254: 165-85.
23 Redner RL, Wang J, Liu J. Chromatin remodelling and leukemia:
new
therapeutic paradigms. Blood 1999;94:417 -28.
24 Fenaux P, Chomienne C, Degos L. Treatment of acute promyelocytic
leu
kaemia. Best Prad Res Clin Haematol2001;14:153- 74.
25 Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, FordJM,
et al. Effi
cacy and safety of a specific inhibitor of the BCR-ABL tyrosine
kinase in
chronic myeloid leukemia. N EnglJ Med 2001;344:1031-7.
26 Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Fernandes Reese
S, Ford JM, et al. Activity ofa specific inhibitor of the BCR-ABL
tyrosine kinase in the blast crisis of chronic myeloid leukemia
and acute lymphoblastic leukemia with the Philadelphia chromosome.
N Engl J Med 2001;344:1038-42.
27 Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov
JP, et al. Molecular classification of cancer: class discovery and
class prediction by gene expression monitoring. Science 1999;286:531-
7.
28 Van Dongen JJM, Seriu T Panzer-Grümayer ER, Biondi A, Pongers
Willemse MJ, Corral L, et al. Prognostic value of minimal residual
disease in childhood acute lymphohlastic leukemia. Lancet 1998;352:
1731-8.
A memorable lecture Lessons of yore
Recently, while working as a senior house officer in a busy emergency
department and only weeks away from giving birth to my first child,
I came across some of my great grandfather's university lecture
notes. He studied medicine at Queen's College in Belfast and later
became a general practitioner in the north of England.
The notes I discovered were beautifully handwritten in black ink
with neat headings and subheadings, all bound together in a black
A5 notebook. In short, the type of lecture notes my peers and I
could only have dreamt of. The subject was "Midwifery,"
and
the first session in the lecture series on pregnancy was taken on
17 October 1901. As I browsed through the pages, I mused on what
antenatal care I would have received if I had been born a century
earlier.
Lecture 15, for instance, informed me of the relevance of food,
clothing, air, stimulants, household conditions, and exercise. The
influence of diet was noted as "very real:' Prochownik of Hamburg
was quoted as advising special diets in the following cases:
"In fat women whose previous labours have been difficult owing
to defective muscular action, by special dieting easier confinements
are procured and the patients are enabled to suckle their children.
"In cases of contracted pelvises (i.e. those with a conjugate
of 31/4 to 4 inches) special dieting will so influence the size,
weight and osseous development of the fetus that normallabour is
obtained:'
(Why did we never hear about this Promownik chap when we were learning
about cephalopelvic disproportion in obstetrics and gynaecology?)
My desire to have lived at the turn of the 19th century culminated
when I read that "the house should be perfect both in ventilation
and sanitation" (I was definitely going to have to have words
with my husband) and that regarding sleep "eight to ten hours
are necessary. Patients should avoid late hours" (I was also
going to have to speak to my consultant about this). When I discovered
that "breasts should be free from all compression and should
be bathed daily with the following lotion: Boracic acid 3i, Whiskey
3i, Water 3iip;' I began to wonder how many women may just have
left out the boracic acid and used the potion for more pleasurable
purposes.
In a later lecture, however, my great grandfather had made slightly
more concerning notes on the aetiology of birthmarks. They can,
he wrote, be explained by "maternal impressions. .. The impression
must be a very strong one-fright etc." This was followed by
an example: "Lady driving saw child run over-over neck. She
was three months pregnant At labour child had scar across neck:'
Having already worked in an inner city emergency department for
several months, where trauma victims were the largest patient group,
I began to wonder whether my unborn child would have any normal
anatomy left I closed the lecture notes and decided it was time
to start my maternity leave.
Lucy James senior house officer in general surgery, Frenchay
Hospital, Bristol
We welcome articles up to 600 words on topics sum as
A memorable patient, A paper that changed my practice, My most
unfortunate mistake, or any other piece conveying instruction,
pathos, or humour. If possible the article should be supplied on
a disk. Permission is needed from the patient or a relative if an
identifiable patient is referred to.
BMJ VOLUME 324 2 FEBRUARY 2002 bmj.com
|