|
MG is Professor of Cell Biology and Director of the Leukaemia Research
Fund Centre
for Cell and Molecular Biology
at the Institute of Cancer Research, London, UK.
Correspondence: Professor Mel Greaves, Institute of Cancer Research,
Chester Beatty Laboratories, 237 Fulham Road,
London SW3 6JB, UK.
Tel: +44 (0) 2078783823.
Fax: +44 (0) 2073520266.
Email: m.greaves@icr.ac.uk
Causal mechanisms in all diseases are diverse and multifactorial,
but medical scientists, as pragmatists, inevitably focus on limited
or circumscribed components of pathogenetic puzzles. In cancer,
epidemiologists have traditionally sought to incriminate exposures;
geneticists uncover inherited susceptibility; and molecular biologists
deconstruct the proximal mechanisms of cell transformation. Molecular
epidemiology promises to deliver new insights in terms of gene-environment
interactions. Each of these endeavours has undeniably provided rich
dividends and insights into cancer causation, but are these likely
to be sufficient as a coherent explanation of our vulnerability
to cancer? I suggest that the biological plausibility of causal
mechanisms would benefit from a historical, evolutionary perspective.
The essential argument is that genes or gene variants and phenotypic
traits that were adaptively selected in the past as advantageous
now contribute crucially to cancer because of their mismatch with
current environmental and social circumstances. The risk attributes
of skin pigmentation and some dietary factors in cancer can be plausibly
interpreted within this context. A case is made here for a Darwinian
perspective on breast and prostate cancers, for which current understanding
of causation is limited and contentious.
Cancers are a collective of some hundreds of cellular disorders
of differing origins and degrees of malignancy, but one essential
feature is shared-the territorial expansion of a mutant clone. Mutations,
accumulated sequentially within descendent subclones over years
or decades, drive the disease via Darwinian or natural selection
of cells ( figure 1) .This process is essentially the same as evolutionary
divergence or serial dominance of species of microbes, plants, and
animals in ecological niches.1,2 The common ingredients are an increase
in numbers by reproduction ( cell proliferation), genetic diversification
within the expanded clone (further gene mutations or deletions in
the stem-cell compartment), environmental pressures and competition
(for tissue space and nutrients), and emergence through these bottlenecks
of anew , dominant subclone. And the process succeeds partly because
the evolution of multicellular creatures, including ourselves as
accidental products, has for some 500 million years and longer retained
the more ancient proclivity of single cells to clone themselves
extensively and emigrate.2 An evolutionary or Darwinian perspective
helps rationalise the inherent risk features of stem cells, the
protracted latency and variable dynamics of cancer, its colonial
tendencies, and its eventual therapeutic intransigence.2 The whole
process comes down to natural selection. In this essay, I advance
the view that the evolutionary principles of Darwinian adaptation
and selection operate also at another level in cancer-in causation.
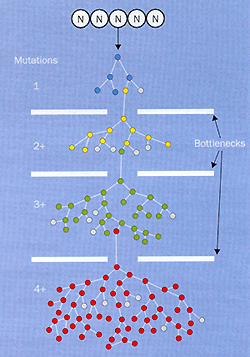
Figure 1. Clonal evolution of a cancer. All cancers evolve
by Darwinian principles: clonal proliferation, genetic diversification
within the clone, and selective pressure enabling mutant subclones
to bridge the bottlenecks (such as anoxia, restricted space and
nutrients, apoptosis imposition). Each colour in the figure represents
a cell (and its descendent clone) acquiring the first (blue) or
additional, sequential mutations. Grey represents dying cells. This
diagram greatly simplifies the extensive genetic diversity, complex
population structure, and highly variable dynamics of cancer clones.
N, normal stem cells.
Mechanistically, mutations that collectively corrupt cellular control
pathways represent the proximate causal mechanism.³ However,
in a broader context the 'causes' of cancer include those exposures,
genetic characteristics, and physiological factors that induce mutations
or influence the probability of mutations. We now have a general
audit of exogenous and endogenous exposures that can lead directly
or indirectly to cancercausing mutations. Included on the list are
ionising radiation, genotoxic chemicals, microbial agents, chronic
inflammation, dietary factors, and hormones.4 In some cases, the
evidence is incontrovertible: the causal connection between tobacco
carcinogens and lung cancer is overwhelmingly the most important
in terms of worldwide burden.7 Other clear-cut examples are the
links between asbestos fibres and mesothelioma and papillomaviruses
and cervical cancer." We are now also beginning to identify
many inherited variations in genes that either directly lead to
cancer as highly penetrant mutants or, much more commonly, indirectly
modify risk of cancer via their influence on the efficiency of the
body's mechanisms for carcinogen detoxification, antimicrobial immune
response, or repair ofDNA damage.89 The Human Genome Project and
its offspring, the Human Cancer Genome Project, will greatly assist
the definition of cancerrisk genotypes and help endorse the plausibility
of candidate exposures.10 But genetics cannot explain it all; cancer
risk can also be modified by other factors, particularly dietary
intake.6lll'
Causal networks
From this cocktail of ingredients, a more comprehensive and plausible
causal network for cancer risk emerges to replace the misleading
concept of a single cause. As in almost all diseases, the causal
mechanism is multifactoriall3 and has components that may operate
in a combinatorial way ( eg, inherited alleles in a susceptible
genotype) .The importance of gene-environment interaction in disease
is now generally recognised.l0 To handle the complexity of cancer
in this way requires merging of disciplines and assembly of predictive
algorithms. It emphasises the importance in causality of the interplay
of exposures, genetic and dietary modifiers, and chance (figure
2). Why chance? Because there is a random element in the way that
DNA gets damaged and leads to mutations; only if the 'right' gene
is mutated or misrepaired in the 'right' way in the 'right' cell
does it matter.'3 And given that our genetic inheritance is itself
a parental lottery, and our environmental exposures may be inadvertent
and invisible, the whole process of cancer is undeniably imbued
with chance, much like evolution in general. This is the way things
work, by and large, in biology.
Armed with adequate data on genetic profiles and relevant exposures,
we might become able to calculate risk of particular cancers for
individuals, although my guess is that such risk estimates will
remain crude or within wide confidence intervals, given the multiplicity
of unquantifiable modifiers of risk and the prevailing influence
of chance.
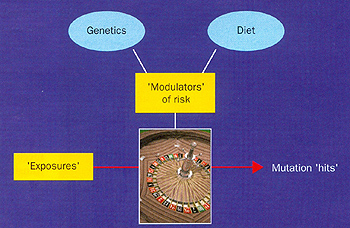
Figure 2. Cancer roulette. Cancer cause is a consequence
of exposures (exogenous or endogenous) that lead to oncogenic mutations
in stem cells in different tissues, but actual risk is always subject
to both modifiers or modulators of risk (principally genetic and
dietary factors) and the ubiquitous role of chance (the roulette
wheel).
The evolutionary angle
Would all of this constitute a satisfactory, sufficient, or complete
explanation of why we get cancer? In a purely mechanistic sense
maybe, but in a wider sense I suggest not. What is missing from
this formulation is a historical dimension, covering both social
and evolutionary time frames. My view is that we cannot make sense
of the apparently high incidence or risk of particular cancers in
particular groups of individuals or societies without reference
to both our evolutionary and more recent historical past.
This argument is part of the perspective of 'evolutionary' or 'Darwinian'
medicine proposed most forcibly and eloquently by physician Randolph
Nesse and evolutionary biologist George Williams.14-1- (For an excellent
recent review see Steams and Ebert.l8) The innovative idea was that
the array of chronic diseases that appears to be associated with
affluence-obesity, adultonset diabetes, cardiovascular disease,
some infectious diseases, allergies, and cancer-has an evolutionary
rationale, as may other medical peculiarities of the human condition,
such as fetal-matemal conflicts, the menopause, and ageing. The
evolutionary arguments are perhaps most well substantiated for pathogen
virulence, resistance to antibiotics, and the emergence ofnew infections.I8I9
For the chronic diseases, including cancer, the underlying concept
is of a mismatch in which our rapid elaboration of social behaviour
patterns has become dislocated from and has outpaced our Stone Age
genetics. If the confrontation is protracted by longevity, eventually,
something has to give.
A key part of this argument rests on the premise that certain normal
(non-mutant) genes and gene variants or alleles selected in the
past because they encoded functions that endowed survival or reproductive
advantage now have the potential indirectly to increase cancer risk
because of a change in the physiological context in which these
same genes are now required to operate. Or, in other words, there
is a nature-nurture mismatch. This twist arises as a paradoxical
consequence of human success at social engineering, engendering
exotic lifestyles driven by social and economic rather than biological
imperatives, and that, together with longer lifespans, teases out
inherent design limitations of our bodies.
The most straightforward and well-rehearsed cancer example is with
skin cancers in pale-skinned caucasians, including both the most
common cancer of all, basal-cell carcinoma, and the most territorially
aggressive of all, melanoma. The essential difference between white
skin and black African skin is in the intracellular amounts and
packaging of melanin pigment. Genetic studies on rodents suggest
that many genes contribute directly or indirectly to skin coloration,
but at some time during the migration of modern human beings northwards
from Africa, there must have been selective pressure favouring paler
skins ( or something linked to paler skin). The reasons for this
development are not entirely clear. The prevailing explanation has
to do with toning down to increase ultraviolet-light-dependent biosynthesis
of vitamin Din cloudy northern regions,20 but whether this is the
whole story is tar from certain.2122 Whatever the exact basis of
selection and the particular genes involved, genetic programming
for paler skins with a diminished melanin ultraviolet filter became
a bonus in survival and reproduction. Now, however, that same advantage
confers a risk of melanoma and other less belligerent skin cancers.
But it is only a serious risk in the context of two other more recent
changes-intermittent, intensive exposure to the sun ( eg, roasting
at noon on Mediterranean beaches) and living long enough for a clone
to accumulate the required set of mutant credentials. Little or
no 'reverse' Darwinian selective pressure can be applied because
most individuals who develop lethal melanoma are past normal reproductive
age. Having said that, however, melanoma now has the fastest rising
rate of any cancer in young adults in some parts of the world, particularly
in northern Europe. Furthermore, the very high frequency of lethal
melanoma in young black Africans with albinism will have exerted
a strong negative selective pressure against transmission of their
genetically altered skin pigmentation.22
Diet and cancer
A broadly similar argument can be made for our genetic adaptations
for dietary intake and metabolism-and the corresponding risk that
contemporary eating habits may harbour for several cancer types.6ll12
Dietary components are complex, and the precise way in which they
contribute to cancer risk, positively or negatively, remains unresolved
and contentious. On the negative side, there is persuasive evidence
that deficiency of foods rich in antioxidants, which protect from
DNA damage, is bad news, especially coupled with excess calorie
intake and the modern trait of a paucity of physical exercise.1223
Many of us now persistently binge, exercise too little, and live
long enough for it to matter. These habits adversely affect the
associated risk of several cancer types, believed to include those
of the breast, prostate, colon, and pancreas, as well as obesity,
adultonset diabetes, and heart disease. The energy surfeit idea
is rendered credible by observations on substantially lower
cancer rates in rodents under calorie restriction2425 and is endorsed
by some epidemiological studies.61 11"3 Potential
mechanisms increasing the probability of malignant transformation
include disruption of insulin signalling networks and persistently
high concentrations of insulinlike growth factor I (IGF I), an inhibitor
of cell death programmes.
This story isn't just about gluttony and sloth; there are also genetic
factors at play. The so-called 'thrifty genotype' provides a plausible,
evolutionary exp1anation of why we have such a problem with these
chronic consequences of overeating and a sedentary existence in
modern societies."' The argument is a deceptively simple one-that
under Stone Age or preagricultural conditions, there might have
been a decided survival advantage (and therefore reproductive advantage)
for individuals whose genetic profile best equipped them for rapid
insulin release and glucose conservation. This pattern would have
accommodated excess intake and storage during transient times of
plenty, as insurance against the likelihood of subsequent famine
or the travails of long-distance migration. Although we do not yet
have adequate insight into the many genes and allelic variants that
are almost certainly involved in this pathway, certain human populations
seemed to be historically well endowed but are now heavily penalised
in this respect-for example, the Arizona Pima Indians, Canadian
Inuits, Australian aborigines, and Polynesians. Once again, earlier
genetic advantage flips to a flaw because the environmental and
behavioural context has changed.
Evolutionary take on breast and prostate cancer
So does the evolutionary argument hold water for any of the other
major types of cancers? My guess is that it makes sense for most
cancers, albeit in varying degrees,2 but let's just consider two
of special interest-those of breast and prostate. The two related
issues here are the generally high risk of these cancers in more
developed societies and the high susceptibility of certain individuals.
Both cancers are very common in North America and northern Europe,
less common in eastern and southern Europe, and even less so in
oriental countries.2~2' Immigrants moving from low-risk countries
to the USA or Australia acquire host-country degrees of risk.~')"'0
These findings alone provide a powerful argument that causation
is linked to lifestyle factors or exposures that are socially or
geographically variable (and therefore potentially preventable)
..j",2Y For breast ( and ovarian]l) cancer, the general view
is that some significant component of risk is attributable to a
chronic consequence of our wholesale deviation from the reproductive
lifestyle of Stone Age women:232 earlier menarche, delayed pregnancy
or none at all,33-35 and lack of protracted breastfeeding.36 Persistent
oestrogenic stimulation could lead to mutational DNA damage indirectly
via sustained ( cyclical) proliferation and endogenous oxidative
stress, via a genotoxic effect of some oestrogen metabolites, or
by a combination of these mechanisms. 'Early' full-term pregnancy
not only provides a break to oestrogenic stress but also may remove
many atrisk cells by terminal differentiation of luminal epithelium.37
The biological plausibility of these associations is supported by
experiments in rodents, in which hormonal mimicry of pregnancy protects
against mammary carcinogenesis.38
At some time during the past 15 million years, females of higher
primate species acquired genetic underpinning of non-seasonal oestrus.
The resultant reproductive traits bequeathed to Homo sapiens are
reflected in contemporary hunter-gatherer tribes, whose serial pregnancies
and protracted breastfeeding habits provide the breaks to otherwise
continuous ovarian cycling for 25 or so years.32 Rates of breast
cancer in such groups are very low. Progressively, however, along
with socioeconomic improvement, emancipation, and contraception,
women have adopted reproductive lifestyles for which, it can be
argued, they are historically and genetically ill adapted. Pump
priming of the ovaries, uterus, and breast in the absence of the
expected outcome of pregnancy continues unabated. Sustained oestrogenic
stress is the result and cancerous mutations a probable consequence.
Hence, the revealing 300-year-old anecdote of Italian nulliparous
nuns (figure 3) whose chosen path of celibacy increases their risk
of breast cancer ,39-41 while protecting them from sexually transmitted
papillomaviruses and cervical cancer .
The effect of these changes seems to be compounded by modern dietary
(and exercise deficit) effects on production of oestrogens and timing
of menarche.2342 A very plausible concept is that the lottery of
who gets breast cancer is largely a combination of modern reproductive
and dietary lifestyles mismatched with genotypic traits that may
have originally conferred benefits, plus the longer time available
for a confrontation, and, of course, chance.
Could the same type of explanation apply to men and their prostates?
There is no unambiguous epidemiological evidence, but a plausible
argument can be made for a composite risk (figure 2) arising in
the evolutionary context. Part of the explanation, given the very
high frequency of occult prostate cancer in both occidental and
oriental ageing men,43 could be that persistent activity of the
prostate incurs cancerous penalty points, provided the man lives
long enough. As far as we know, the prostate gland developed for
one thing only-lubrication of sperm passage. The evolutionary rationale
of the prostate operating at full potential is unclear, but there
may be an adaptive advantage of persistent pump priming ( via testosterone)
of the prostate to optimise chances of successful fertilisation
of females with non-seasonal, covert oestrus.2 This idea might help
explain why male human beings have the biggest prostate gland among
mammalsalong with the domesticated dog, tellingly perhaps the only
other mammal known to suffer from spontaneous prostatic carcmoma.
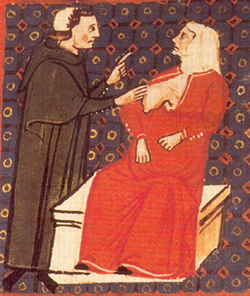
Figure 3. Nuns and breast cancer. Examination of the
breast by the surgeon Teodorico Borgognoni (1275). An apparent excess
of breast cancer among nuns was recognised long ago in Europe. The
pioneering Italian epidemiologist Bernadino Ramazzini was perhaps
the first to draw attention to this excess in his descriptions of
lifestyle, occupation, and disease,394o along with his memorable
quote, "You can seldom find a convent that does not harbour
this accursed pest, cancer, within its walls". More systematic
information on the risks of breast and cervical cancers in nuns
was provided by Rigoni-Stern in his survey of cancer deaths in Verona,
1760-1839.239 Given the date of the above painting (1275), breast
cancer has probably been common in nuns for many
centuries. The painting is from Leiden University (MS Vossios Lat
F 3, fol 90v) and has been published previously in a review of the
history of breast cancer.41
This cannot be the whole story, however, as the internationally
varied incidence rates and the influence of migration testify. There
must also be an explanation ofwhy age-specific rates of prostate
cancer have apparently increased in the past few decades, independently
of screening, which has certainly inflated reported incidence rates,
especially in the USA.44 Some feature of modern or western lifestyle,
as with breast cancer, may, in combination with longevity, be exacerbating
a nature-nurture mismatch. Epidemiological studies have yet to provide
clear evidence, but one component again may be diet-Iow intake of
antioxidants or phytooestrogens, perhaps. There is some evidence
from experimental prostate carcinogenesis in mice that the polyphenolic
constituents of green tea are protective.45 An unfavourable ratio
of diet to exercise, raising concentrations of IGFl or affecting
the testosterone signalling pathway may also be important.46
Another factor promoting the transition of incipient to full-blown
cancer could be sexual activity itself, recycling stress in the
prostate.2 Male human beings may have been adaptively primed in
our early evolutionary history for persistent prostate activity,
but persistent sexual activity keeping the prostate in business
after 'normal' reproductive age ( ie, over age 50 years) is biologically
exotic behaviour, albeit very common and natural in modern societies.
Needless to say, this activity itself depends on the continued efficacy
of testosterone signalling, and therefore sexual activity could
be a surrogate marker rather than a cause of prostate stress. Epidemiological
evidence bearing directly on this idea is sparse and ambiguous.
Studies on the risk of prostate cancer in celibate Catholic priests,
for example, have produced mixed results.47 In a study of sexual-behaviour
variables and prostate-cancer risk, there was no overall association
with number of partners or frequency of intercourse, but there was
an unpredicted (by the researchers) observation of a significant
trend towards increased risk with greater frequency of intercourse-for
men in their fifties, but not at younger ages.48 This uncomfortable
issue has not been adequately investigated.
All in the genes?
If these ideas are correct, they might be endorsed by genetic studies.
Such studies might also tease out the difference between the general
susceptibility that exists at population level in modern western
societies as a consequence of behavioural changes from the increased
susceptibility inherited by some individuals but not others. We
might expect, for example, that inherited genetic polymorphisms
that impinge on the oestrogenic and androgenic signalling pathways,
and perhaps on insulin circuitry also, would confer altered risk
of breast and prostate cancer for certain individuals in excess
of the risk derived from the nature-nurture mismatch referred to
earlier.
Constitutive genetic factors clearly do contribute very significantly
to risks of breast and prostate cancers, around 10% of cases occurring
in a familial pattern.49 This clustering has been attributed to
the inheritance of dominant or highly penetrant mutant genes including
BRCAl and BRCA2. Inheritance of such genes can increase breast-cancer
risk up to a 50-80% probability. These dangerous genes have origins,
as mutant varieties, hundreds or a few thousand years ago. The most
plausible explanation for the high prevalence of individual BRCAl
or BRCA2 mutations in certain populations ( eg, Ashkenazi Jews50)
is not via any adaptive or 'useful' selection but via the historical
founder effect-ie, migrant colonisation of an isolated geographic
region by a very small number of people including one individual
in whose germ cell the mutation first arose.5l As long as there
was no adverse effect on reproductive fitness, there would be no
strong selective pressure against mutation carriers (female or male),
provided that carriers reproduced before they developed cancer and
died.
Other studies suggest, however, that the prevalence of familial
pedigrees of breast and prostate cancers underestimates the contribution
that inherited genes contribute to risk, not only because new mutant
genes can arise in the germline at each generation. Some insight
into the contribution of inherited genetics to cancer risk or that
of any other disease can be deduced from comparison of frequency
or concordance of disease in identical or monozygotic twins versus
dizygotic (fraternal) twins. This type of analysis, introduced by
Darwin's cousin Francis Galton 125 years ago, has proved very productive
in medicine, though interpretation of data is not without difficulty.
In the largest analysis of this type for adult cancer, involving
a cohort of 44 788 Scandinavian twin pairs, the outcome varied with
cancer type.52 For lung cancer, monozygotic twins showed very little,
if any, increase in concordance over dizygotic twins, whereas for
prostate and breast cancers, the differences were very large. Although
these data are subject to some limitations,5354 they suggest that
around 40% of prostate-cancer risk and just under 30% of breast-cancer
risk can be ascribed to inherited susceptibility. Analysis of risk
of contralateral breast cancer and risk in twins has provocatively
suggested that most cases of breast cancer may have an important
genetic risk component.55
The general interpretation of these data is not, or should not be,
that breast and prostate cancers are deterministic genetic diseases
like cystic fibrosis or betathalassaemia. Neither is it very likely
that the one in ten women in more developed countries at risk of
breast cancer over their lifetime were all born harbouring silent
mutant genes lurking with malign intent. A more likely explanation
is that some normal and common variants ot certain genes can, with
moderate probabilities, amplify still further the increased risk
of these cancers-ie, a racheting up of the gene-environment mismatch.
Several inherited gene variants probably have to operate in concert
to constitute a risky genotype. If this interpretation is correct,
we face a conundrum-why are risky genotypes tor breast and prostate
cancers apparently so common?
Genetic susceptibility: winners become losers?
The explanation may well lie in some part of gene-environment-behavioural
interactions with evolutionary overtones. It will doubtless become
clearer once we identify all the genes involved and what the proteins
they encode actually do, hence the relevance to cancer of the Human
Genome Project. To date, studies of candidate gene polymorphisms
in breast cancer have yielded somewhat equivocal or inconsistent
results.56,57 Arguably the most informative molecular epidemiological
data so far for prostate and breast cancers implicate variants in
genes with products involved in the sequential synthesis, metabolism,
or cellular receptor binding and signalling of androgens and oestrogens.58
64 The evidence currently is more persuasive for prostate than for
breast cancer. Significantly, most, if not all, alleles that seem
to be associated with increased risk of prostate or breast cancer
are known or predicted to encode protein functions that increase
activity in the hormonal signalling pathway.58-64 Several, though
not all, of these alleles are very common and are more prevalent
in ethnic groups with well recognised high risks ( eg, African Americans
and prostate cancer).65 Some alleles may be associated with advanced
rather than localised disease, thus underpinning late, promotional
events.64 All cell signalling networks are regulated by positive
as well as negative controls; short polymorphic polyglutalline repeats
of the androgen receptor, which increase receptor signalling and
prostatecancer risk,61 have the opposite effect in postmenopausal
women of decreasing risk of breast cancer, possibly by counteracting
oestrogenic signalling.62 In this context, another relevant finding
is that the BRCAl gene, which is mutationally inactivated (in one
copy) in carriers, encodes a protein that normally represses transcription
of the oestrogen receptor.66 The breast-cancer risk in women who
carry BRCAl or BRCA2 is high but variable and may itself be modified
by coinheritance of polymorphic alleles at other loci. Rebbeck and
colleagues found that risk was increased by the presence of long
polyglutalline repeats in a gene, AIBI, that increases oestrogenic
transcriptional signalling in the mammary gland.67
Altogether, these data clearly point in the direction of genetic
control of sex-hormone signalling contributing to variation in risk
susceptibility. Critics may argue that these preliminary genetic
susceptibility screens with individual candidate genes have given
low odds ratios for risks and, in some cases, inconsistent results.
That is hardly surprising, particularly since what is likely to
be critical is the genotypic variation underpinning a complete signalling
pathway or network rather than any individual component. Neither
should we expect that only one pathway influences risk of mutational
changes in prostate or breast stem cells. Alleles affecting oxidative-stress
or DNA-repair pathways are likely to contribute to risk. Vitamin
D influences proliferation and apoptosis of prostatic cells, and
preliminary genetic data implicate alleles of the vitamin D receptor
in risk of advanced prostate cancer .64
Why now would such genotypes constitute an increased risk for cancer?
In a superficial sense, the answer is obvious: many, if not all,
breast and prostate cancers start their own evolutionary journey
being driven by oestrogen and testosterone, respectively. Further,
hormone- independent, progression of prostate cancers may be facilitated
by ligand-independent triggering of amplified androgen receptors.68
As human beings pass the 5O-year barrier, sex hormone concentrations
decline, and the idea that those of us with the more persistently
increased hormonal signalling are most at risk seems to make sense.
This idea fits with the concept of sustained hormonal stress and
accords with the experimental data on tullours induced in animals
by long-term administration of sex hormones69,70 and with the anecdotal
evidence of breast -cancer development in trans-sexual men injected
with high doses of oestrogens.71
These preliminary genetic data and arguments still beg the question
of the historical or evolutionary rationale for the apparently high
prevalence of risky genotypes. Stephen Gould and some other prominent
evolutionary biologists are sceptical of ascribing everything that
exists in the biological world to previous adaptive advantage and
selection, and any such supposition does require exallination.72
Nevertheless, a Darwinian interpretation would make sense here,
although it would be difficult to substantiate. The speculative
argument I advance is that selective advantage in our historical
past accrued from chance acquisition of gene variants that endowed
substantial metabolic advantages ( under prevailing environmental
conditions)-vitallin D synthesis, energy storage, or greater reproductive
success. Nothing favours Darwinian selection more than survivability
and being out front in the mating game.
Women genetically well endowed with enhanced fertility, coupled
perhaps with efficient energy storage capacity ( via alleles in
insulin circuitry and fat metabolism), would undoubtedly have in
past times been at a distinct advantage in terms of passing on their
genes. Men with gene variants facilitating more effective prostate
priming coupled perhaps not only with fertility, but with mate-attracting
potency, hunting ability, and survivability, might be expected to
accrue reproductive advantage to be passed on to their descendants.
But in contemporary societies, these same beneficiaries may be those
most at increased risk of cancer via persistent proliferative activity
of breast or prostate stem cells, oxidative stress, and DNA damage,
particularly now that lifespans are longer . Evolutionary counter-pressures
can exert little or no influence if the major downside or trade-off
( cancer) arises in predominantly ageing and reproductively inactive
individuals. The argument is not that all human genes or alleles
that are associated with increased cancer risk have a historical
rationale in adaptive selection, but that many such genes will have
this rationale.
We will not have a wholly satisfactory grasp of the causal mechanisms
for breast and prostate cancer until we fully appreciate that cause,
in reality, is an interactive network of proximal and distal events.
We need to improve in developing algorithms that compute the compound
risk derived from genotypic variation, diet, reproductive physiology
, and behaviour, and allow for the ubiquitous role of chance. The
complex interaction of our unique genotypes and our natural or concocted
environments and lifestyles dictates risk of many cancers. How this
risk translates into actual rates of disease that vary in time and
place may begin to make sense only in the context of our evolutionary
and social history.
Search strategy and selection criteria
Relevant publications (up to the end of 2001) were screened by systematic
review of the major cancer-research journals (both review journals
and those with experimental papers). A PubMed search was used with
the key words "Darwinian cancer", "breast cancer,
evolution", and "prostate cancer, evolution". Papers
were selected on the criteria of direct relevance, a personal judgment
on the quality of study design, and strength of conclusion. In some
cases, review articles are quoted for convenience rather than primary
data sources.
Acknowledgments
I thank the Institute of Cancer Research and Leukaemia Research
Fund for their support, Alan Ashworth, Mitch Dowsett, Jill Ross,
Randy Nesse, Freda Alexander, and Barbara Deverson and Chris Priest
for help in preparing the paper.
References
1 Nowell PC. The clonal evolution of tumor cell populations.
Science 1976; 194: 23-28.
2 Greaves M. Cancer: the evolutionary legacy. Oxford: Oxford
University Press, 2000.
3 Hanahan D, Weinberg RA. The hallmarks of cancer.
Cell 2000;lOO: 57-70.
4 Doll R, Peto R. The causes of cancer. Oxford: Oxford University
Press,1981.
5 Schottenfeld D, Fraumeni JF, (eds). Cancer epidemiology and prevention,
211d ed. New York: Oxford University Press, 1996.
6 Peto J. Cancer epidemiology in the last century and the next decade.
Nature 2001; 411: 390-95.
7 Peto R, Chen ZM, Boreham J. Tobacco: the growing epidemic.
NatMed 1999; 5: 15-17.
8 Perera FP. Molecular epidemiology: on the path to prevention?
J N atl Cancer I nst 2000; 92: 602-12.
9 Ponder BAJ. Cancer genetics.
Nature 2001; 411: 336-41.
10 Clayton D, McKeigue PM. Epidemiological methods for studying
genes and environmental factors in complex diseases.
Lancet 2001 ;
358: 1356-60.
11 World Cancer Research Fund in association with the American Institute
for Cancer Research.
Food, nutrition and the prevention of cancer: a global perspective.
Washington:
American Institute for Cancer Research, 1997.
12 Griffiths K, Adlercreutz H, Boyle P, et al. Nutrition and cancer.
Oxford: Isis Medical Media, 1996.
13 Dubos R. Mirage ofhealth. New York: Harper and Brothers, 1959.
14 Nesse R, Williams G. Evolution and health: the new science of
Darwinian medicine. London: Weidenfeld & Nicolson, 1995.
15 Nesse RM, Williams GC. Evolution and the origins of disease.
Sci Am 1998; November: 58-65.
16 Stearns SC, (ed). Evolution in health and disease. Oxford: Oxford
University Press, 1999.
17 Trevathan WR, Smith EO, McKenna JJ, (eds). Evolutionary
medicine. New York: Oxford University Press, 1999.
18 Stearns SC, Ebert D. Evolution in health and disease: work in
progress.
Quart Rev Biol2001; 76: 417-32.
19 Ewald PW. Evolution of infectious diseases. Oxford: Oxford
University Press, 1994.
20 Loomis WF. Skin pigmentation regulation of vitamin-D biosynthesis
in man.
Science 1967; 157: 501-06.
21 Diamond J. The rise and fall of the third chimpanzee. UK: Radi
us
Publishing, 1991.
22 Rees J. Post-genome integrative biology: so that's what they
call clinical science.
Clin Med 2001; I: 393-400.
23 Mezzetti M, La Vecchia C, Decarli A, et al. Population attributable
risk of breast cancer: diet, nutrition, and physical exercise.
J Natl Cancer Inst 1998; 90: 389-94.
24 Cohen LA, Choi KW, Wang CX. Influence of dietary fat, caloric
restriction,
and voluntary exercise on N-nitrosomethylureainduced mammary tumorigenesis
in rats.
Cancer Res 1988; 48: 4276-83.
25 Grasl-Kraupp B, Bursch W, Ruttkay-Nedecky B, Wagner A, Lauer
B, Schulte-Hermann R. Food restriction eliminates preneoplastic
cells through apoptosis and antagonizes carcinogenesis in rat liver
.
Proc Natl Acad Sci USA 1994; 91: 9995-99.
26 Neel JV, Weden AB, Julius S. Type II diabetes, essential hypertension,
and obesity as "syndromes of impaired genetic homeostasis":
the "thrifty genotype" hypothesis enters the 21 st century.
Perspect Biol Med 1998; 42: 44-74.
27 Parkin DM, Pisani P, Ferlay J. Estimates of the world wide incidence
of eighteen major cancers in 1985. IntJ Cancer 1993; 54: 594-606.
28 Dhom G. Epidemiology ofhormone-depending tumors. In: Voigt KD,
Knabbe C, (eds). Endocrine dependent tumors.
New York: Raven Press, 1991: 1-41.
29 Parkin DM. Global cancer statistics in the year 2000.
Lancet Oncol 2001; 2: 533-43.
30 Bouker KB, Hilakivi-Clarke L. Genistein: does it prevent or
promote breast cancer?
Environ Health Perspect 2000; 108: 701-08.
31 Risch HA. Hormonal etiology of epithelial ovarian cancer, with
a hypothesis concerning the role of androgens and progesterone.
J N atl Cancer I nst 1998; 90: 1774-86.
32 Eaton SB, Pike MC, Short RV, et al. Women's reproductive
cancers in evolutionary context. Quart Rev Biol1994; 69: 353-67.
33 Henderson BE, Ross RK, Pike MC, Casagrande JT. Endogenous
hormones as a major factor in human cancer.
Cancer Res 1982; 42:3232-39.
34 Lipworth L. Epidemiology of breast cancer.
Eur J Cancer Prev 1995; 4: 7-30.
35 Wohlfahrt J, Melbye M. Age at any birth is associated with breast
cancer risk. Epidemiology 2001; 12: 68-73.
36 Labbok MH. Effects of breastfeeding on the mother.
Pediatr ClinNorth Am 2001; 48: 143.
37 Russo J, Gusterson BA, Rogers AE, et al. Biology ofdisease:
comparative study of human and rat mammary tumorigenesis.
Lab Invest 1990; 62: 244-78.
38 Guzman RC, Yang J, Rajkumar L, et al. Hormonal prevention of
breast cancer: mimicking the protective effect of pregnancy. Proc
Natl Acad Sci USA 1999; 96: 2520-25.
39 Mustacchi P. Ramazzini and Rigoni-Stern on parity and breast
cancer. Arch Intern Med 1961; 108: 639-42.
40 Ramazzini B. Diseases of workers. Translated from the Latin text
of
1713 by Wilmer Cave Wright. New York: Hafner Publishing Co,
1964.
41 DeMoulin D. A short history of breast cancer. Dordrecht: Kluwer
Publishers, 1983.
42 Coffey DS. Similarities of prostate and breast cancer: evolution,
diet, and estrogens.
Urology 2001; 57: 31-38.
43 Breslow N, Chan CW, Dhom G. Latent carcinoma of prostate at autopsy
in seven areas.
Int J Cancer 1977; 20: 680-88.
44 Hsing A W , Tsao L, Devesa SS. International trends and patterns
of prostate cancer incidence and mortality.
Int J Cancer 2000; 85:60-67.
45 Gupta S, Hastak K, Ahmad N, et al. Inhibition of prostate carcinogenesis
in TRAMP mice by oral infusion of green tea
polyphenols. Proc Natl Acad Sci USA 2001; 98: 10350-55.
46 Cohen P. Serum insulin-like growth factor-I levels and prostate
cancer risk: interpreting the evidence.
J Natl Cancer Inst 1998; 90:876-79.
47 Kaplan SD. Retrospective cohort mortality study of Roman
Catholic priests. Prev Med 1988; 17: 335-43.
48 Hayes RB, Pottern LM, Strickler H, et al. Sexual behaviour, STDs
and risk for prostate cancer. Br J Cancer 2000; 82: 718-25.
49 Szabo CI, King M -C. Population genetics of BRCA I and BRCA2.
Am J Hum Genet 1997; 60: 1013-20.
50 Roa BB, Boyd AA, Volcik K, Richards CS. Ashkenazi Jewish population
frequencies for common mutations in BRCAl and
BRCA2. Nat Genet 1996; 14: 185-87.
51 Diamond JM, Rotter JI. Observing the founder effect in human
evolution. Nature 1987; 329: 105-06.
52 Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and
heritable factors in the causation of cancer: analyses of cohorts
of twins from Sweden, Denmark, and Finland. N Engl J Med 2000; 343:
78-85.
53 Hoover RN. Cancer: nature, nurture, or both. N Engl J Med 2000;
343: 135-36.
54 Risch N. The genetic epidemiology of cancer: interpreting family
and twin studies and their implications for molecular genetic
approaches. Cancer Epidemiol Biomarkers Prev 2001; 10: 733-41.
55 Peto J, Mack TM. High constant incidence in twins and other relatives
ofwomen with breast cancer. Nat Genet 2000; 26:
411-14.
56 Dunning AM, Healey CS, Pharoah PDP, et al. A systematic review
of genetic polymorphisms and breast cancer risk. Cancer
Epidemiol Biomarkers Prev 1999; 8: 843-54.
57 Clemons M, Goss P. Estrogen and the risk of breast cancer. N
Engl
JMed2001; 344: 276-85.
58 Ross RK, Pike MC, Coetzee GA, et al. Androgen metabolism and
prostate cancer: establishing a model of genetic susceptibility.
Cancer Res 1998; 58: 4497-504.
59 Feigelson HS, McKean-Cowdin R, Coetzee GA, et al. Building a
multigenic model of breast cancer susceptibility: CYP17 and HSDl7Bl
are two important candidates. Cancer Res 2001; 61: 785-89.
60 Habuchi T, Liqing Z, Suzuki T, et al. I ncreased risk of prostate
cancer and benign prostatic hyperplasia associated with a CYP17gene
polymorphism with a gene dosage effect. Cancer Res 2000; 60: 5710-13.
61 Stanford JL, Just JJ, Gibbs M, et al. Polymorphic repeats in
the androgen receptor gene: molecular markers of prostate cancer
risk. Cancer Res 1997; 57: 1194-98.
62 Giguere Y, Dewailly E, Brisson J, et al. Short polyglutamine
tracts in the androgen receptor are protective against breast cancer
in the general population. Cancer Res 2001; 61: 5869-74.
63 Makridakis NM, Ross RK, Pike MC, et al. Association of mis-sense
substitution in SRDSA2 gene with prostate cancer in AfricanAmerican
and Hispanic men in Los Angeles, USA. Lancet 1999; 354: 975-78.
64 Ingles SA, Ross RK, Yu MC, et al. Association of prostate cancer
risk with genetic polymorphisms in vitamin D receptor and androgen
receptor. J Natl Cancer Inst 1997; 89: 166-70.
65 Shibata A, Whittemore AS. Genetic predisposition to prostate
cancer: possible explanations for ethnic differences in risk. Prostate
1997; 32:
65-72.
66 Zheng L, Annab LA, Afshari CA, et al. BRCA1 mediates ligand independent
transcriptional repression of the estrogen receptor . ProcNatlAcad
Sci USA 2001; 98: 9587-92.
67 Rebbeck TR, Wang Y, Kantoff PW, et al. Modification of BRCA land
BRCA2-associated breast cancer risk by AIBI genotype and reproductive
history. Cancer Res 2001; 61: 5420-24.
68 Visakorpi T, Hyytinen E, Koivisto P, et al. In vivo amplification
of the androgen receptor gene and progression ofhuman prostate cancer.
Nat Genet 1995; 9: 401-06.
69 Noble RL. The development of prostatic adenocarcinoma in Nb rats
following prolonged sex hormone administration. Cancer Res 1977;
37: 1929-33.
70 Kwapiem RP, Giles RC, Geil RG, Casey HW. Malignant mammary tumors
in beagle dogs dosed with investigational oral contraceptive steroids.
J Natl Cancer Inst 1980; 65: 137-42.
71 Symmers WSC. Carcinoma of breast in trans-sexual individuals
after surgical and hormonal interference with the primary and secondary
sex characteristics. BM] 1968; 2: 83-85.
72 Rose MR, Lauder GV, (eds). Adaptation. San Diego: Academic
Press, 1996.
|