G. F. Pluzhnikova 1, 0. V. Plutalov3, U. A. Berlin 3, and E. I. Scwartz2 H鄝atol. Bluttransf. Vol 35 |
|
1. The N. N. Petrov Research Institute of On cology,
USSR Ministry of Health, Lenin grad, USSR.
It is widely accepted that tumor development and progression are due to illegitimate activation of cellular oncogenes by point mutation, retroviral insertion, chromosomal translocation and amplification or deletion of the genc [1- 3]. Nonrandom deletions of chromosomal regions 13q14 and 11p13 have been detected in retinoblastoma [1] and Wilm's tumor [2, 3]. It has been proposed that these rare childhood cancers result from the deletion of dominant-acting genes, permitting the expression of tumorigenic recessive alleles [1 ]. Moreover, restriction fragment length polymorphism (RFLP) analysis has demonstrated loss of H-ras.I oncogene allele (chromosome 11 p 15) in primary bladder, breast, ovarian, and lung carcinomas [4- 7]. On the other hand, another important mechanism of activation of ra.s oncogene (including H-rasI) have been shown in 10-15% of certain types of human tumors, which involved a point mutation, causing an alteration at amino acid positions 12, 13 or 61 of the ras gene product p21 ras [8]. The study discusses the possible supressive action of the wild-type H-ras.I allele on the mutant one in breast cancer.
The RFLP of H-rasI oncogene was analyzed in 76 primary breast carcinomas
as described [7]. H-ra.sI sequence spanning 145 base pairs across
cod on 12 was amplified in vitro by Thermu.s thermophyfus DNA polymerase
[9]. Subscquent MspI digestion allowed us to detect the mutation
in "hot spot" due to the loss of the restriction site for MspI in
the case of substitution in the 12th codon of H-rasI [10]. 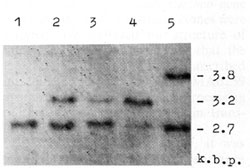 Fig. I. Deletions of one of H-rasI allele in breast carcinomas (BC), identified by means of PvuII restriction of DNA samples, Southern blotting and hybridization with 6,6-fragment of pEl [II]. Samples of DNA were derived from (I) BCI2 -genotype A1/A1; (2) leukocytes of BC9 constitutive genotype A 1/A2; (3) BC9 A1/A2, deletion of A2 allele; (4) BC 5 -A 1/ A 2, deletion of A 1 allele; and (5) BC 31 genotype A 1/ A3. Slight hybridization signals at the place of lost A 2 (3) and A 1 (4) alleles are due to the contamination of the tumors by normal cells. In BC 107 and BC 109 the same deletions were detected as in BC9 (3)
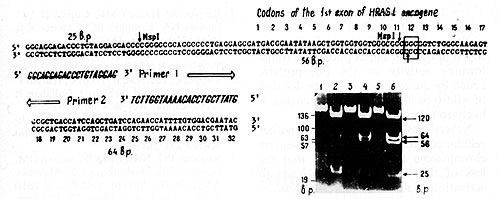 Fig.2. Detection of H-rasI mutation in codon 12. The 145 base pair (bp) DNA sequence across 12th codon (in the frame) was amplified in vitro using the oligonucleotide primers (large open arrows indicate the 5' 3' orientation of the primers [9]. MspI digestion of the amplified sequences result in three fragments (25, 56, and 64 bp) in the case of wild-type allele, and two fragments (25 and 120 bp) in the case of substitution in codon 12 that altered msp site of restriction (vertical arrows). The photograph demonstrates the products of amplification of BC5 (1), BC9 (3), and BC107, BC109 (not shown) DNA samples and MspI restriction (3, 4, and 6, respectively). Arrows on the right of the photograph indicate bands corresponding to mutant and wild-type alleles. Alu fragments of pBR322 DNA (standard) are pointed out on the left of the photo
Restriction analyses of 76 DNA samples from primary breast carcinomas
revealed deletions of one of the H -rasI allele in ten out of 41
(25% ) heterozygous patients (Fig.1). Enzymatic amplification and
Msp restriction showed the presence of point mutation in the undeleted
H-rasl allele in four out of ten carcinomas with the allelic loss
(Fig. 2). All four mutations identified were the G-to- T transversion
in the second position of cod on 12 (Fig. 3). 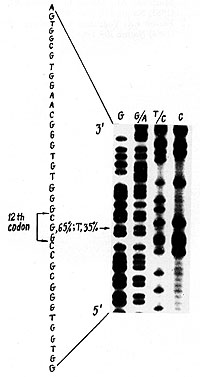
The loss of intact H-rasI allele and consequently its product -p21 ras -nor mally involved in transport of mitogenic signal in the cell, might potentiate transforming activity of the oncoprotein, coded by the mutant allele. The deletion of wild-type allele of H-rasI oncogene is likely to unmask the mutant one. Nevertheless it is possible that another cellular constraint of growth is present on chromosome 11 p 13-p 15, and that the loss of this suppressor locus leads to activation of normally repressed class of genes.
1. Cavenee WK, Dryja TP, Phillips RA, Benedict WF, Godbourt R,
Gallie BL, Murphree AL, Strong LC, White RL (1983) Nature 305:779-784
|