|
1 Supported by a Fellowship of the National Cancer
Cytology Center and a Fellowship of the Deutsche F orschungsgemeinschaft
2 Supported by Grants POI CA-19266, ROl CA 22677 and ROl CA-37l56
from the National Cancer Institute Department of Pathology,
University of Chicago, Chicago, IL 60637, USA
A. Introduction
Transplantation experiments have clearly demonstrated the existence
of unique (individual) tumor-specific antigens on cancers induced
by physical or chemical carcinogens. These antigens often induce
a tumor-specific immune response upon immunization with a tumor
which protects the host against a subsequent challenge with the
same tumor, but not against a challenge with any other independently
induced tumor [1]. Unique antigens were observed even when the tumors
were induced with the same carcinogen in the same organ system in
the same strain of mice [2]. This finding of unique tumor specificity
raises questions about the mechanism by which these tumorspecific
antigens are generated. The critical questions regarding such unique
tumor-specific antigens are their composition, genetic origin, and
possible role as target antigens for the immune system. However,
the identification of tumor-specific antigens that cause tumor rejection
has proven to be extremely difficult in the past. Serological probes
with unique tumor specificity are difficult to obtain [3], and the
serologically recognized antigens may not be the target for tumor
rejection [4] that is primarily T -cell mediated [5]. We used UV-induced
tumors of mice for studying the nature of tumor-specific antigens
for the following reasons: (a) the unique tumor-specific rejection
antigens on UV -induced tumors are stronger than those on chemically
induced tumors, in that UVinduced tumors often regress after transplantation
into normal mice even without prior immunization; (b) several of
the tumor-specific rejection antigens on one such UV-induced regressor
tumor, called 1591RE, have been defined by cytolytic T -cell clones;
(c) monoclonal antibodies with unique specificity for this UV-induced
regressor tumor have been generated which reacted with novel MHC
class 1 molecules on this tumor; and (d) the genes encoding the
antibody-recognized novel class 1 molecules have been cloned and
identified by transfection. We describe here the relationship between
the novel MHC class I molecules encoded by the cloned genes and
the rejection antigens of the 1591 tumor. Recently, we found that
one of the novel 1591 class I genes encodes an antigen that causes
immunological tumor rejection in normal mice [6]. Transfection of
this novel class I gene into a 1591 progressor tumor variant leads
to the rejection of the gene-transfected progressor tumor, demonstrating
that a single gene can revert the progressive growth behavior and
establish the regressor phenotype characteristic of the parental
1591-RE tumor .
B. Results
The 1591 tumor contains three novel class I genes designated 216,166,
and 149 which account for the abnormal reactivity of the tumor cells
with MHC class I-specific monoclonal antibodies [7]. The gene 216
encodes an antigen that is selectively recognized by the 1591 tumor-specific
antibody CP28 [8]. The molecules encoded by the genes 149 and 166
cross-react with monoclonal antibodies specific for allogeneic MHC
class I antigens. Together, the three 1591 class I genes 216, 166,
and 149 can account for all the novel MHC class I determinants expressed
by


1591 tumor cells (Fig.1). In addition, the 1591-RE1 tumor expresses
multiple independent CTL-defined antigens [9] each of which can
independently cause tumor rejection.In the first part ofour study,
we determined whether any of the three novel MHC class I genes (216,
166, or 149) encoded the antigen recognized by anti-A CTL. We have
shown previously that tumor variants selected for the loss of the
anti-A CTL-defined antigen are no longer rejected by normal mice
[10] implicating a close linkage between ( or even identity of)
the A antigen and the antigen leading to tumor rejection. However,
careful attempts to block A antigenspecific CTL clones with antibodies
specific for anyone of the three novel class I MHC antigens encoded
by the 216, 166, or 149 genes failed [11]. Therefore, the relationship
of the serologically defined novel class 1 antigens to the CTL-defined
antigen remained uncertain. We transfected the novel 1591 class
1 genes into mouse L cells and used these gene-transfected cells
as targets for the 1591 tumor-specific CTL lines. Figure 2 shows
that only the 216 gene-transfected L cell line was killed by the
anti-A CTL line while L cells transfected with the 166 gene or the
149 gene were not affected by the anti-A CTL clone. The A -B + C
-D- or the A -B-C-D- variants of the 1591 were not killed. As expected,
however, the A + B C-D- variant of 1591 was killed by the antiA
CTL, as was the A+B+C-D- parental 1591-RE regressor tumor line.
Anti-B, antiC, or anti-D CTL did not kill any of the L cells transfected
with the novel MHC class I genes (not shown). Together, our data
clearly indicate that the 216 gene-encoded novel class I antigen
is recognized by both the CP28 monoclonal antibody (Fig. 1) as well
as the anti-A CTL clone (Fig. 2). We have shown previously that
all the in vivo- or in vitro-derived progressor variants of the
1591-RE tumor had lost all three novel class 1 genes 216, 166, and
149 simultaneously [6]. It was not clear whether the presence of
the 216 gene would alone be sufficient to establish the regressor
phenotype. To test this, we introduced by transfection the 216 gene
into a progressively growing A- 1591 variant, designated 1591-PRO,
which had lost all three novel class I genes. This progressor tumor
was cotransfected with the 216 gene and the gene encoding the enzyme
aminoglycoside phospho transferase which confers resistance to the
drug 0418. The 0418-resistant cell population was cloned and 24
of77 clones expressed the 216 gene-encoded antigen as determined
by their reactivity with the CP28 antibody. Two 216 genes expressing,
A antigen-positive clones, designated TR216+.1 and TR216+.2, and
two negative clones, designated TR216-.3
Table 1. Reversal of malignant growth
in normal mice by transfection of the novel class I gene 216

a
A clone of the progressor tumor, 1591-PRO was transfected with the
216 gene and the neomycinresistant gene. The G418 drug-resistant cell
population was cloned and two clones which expressed the 216 gene-encoded
antigen (1591-PRO TR216+1 and 1591-PRO TR216+2) and two clones which
did not express the 216 gene-encoded antigen (1591-PRO TR216-3 and
1591-PRO TR216 -: 4) were used to challenge five normal mice or two
nude mice with tumor fragments con taining > 10 high 8 tumor cells.
b
Expression of the 216 gene product was de termined by F ACS IVB analysis
using the monoclonal antibody CP28 that specifically recognized this
gene product and a fluoresceinated second antibody. Cell lines designated
positive for expression of the 216 gene product stained at least two
times above background (binding ratio > 2), while all cell lines designated
negative for 216 gene expression stained less than 1.5-fold above
back ground (binding ratio < 1.5).
c
Number of mice with progressively growing tumors/number of mice challenged.
Mice receiving the 216- clones died within approximately 6 weeks due
to the large tumor burden. The mice that were challenged with the
216 + transfectants did not develop tumors even after 4 months, except
for one mouse that grew out an antigen loss variant approximately
2.5 weeks after injection. AIl cell lines used in this experiment
readily formed tumors in nude mice.
d
One of the mice injected with the transfected 1591-PRO TR216+.1 cell
line developed a progressively growing tumor that was reisolated (designated
1591-PRO TR216.1 reisolate) and reanalyzed for expression of the 216
gene by F ACS IVB (Fig. 3) and for tumor incidence in normal mice.
(Reproduced from Ref.6)
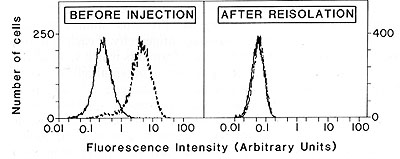
Fig.3. Loss of expression of the transfected 216 gene
in a reisolated 1591-PRO tumor that grew progressively despite transfection
with the 216 gene. The 216 gene-transfected tumors analyzed are
the same as those used for the experiments described in Table 1.
Thefirst panel shows the histogram of the 1591-PRO TR216+.1 cell
line and the second panel shows the histogram of the 1591-PRO TR216.1
reisolate. Cells were incubated with the monoclonal antibody CP28
followed by incubation with fluerescein-coupled goat anti-mouse
immunoglobulin antibodies (--) or incubated with only the goat anti-mouse
immunoglobulin (-) Ten thousand cells were analyzed with the F ACS
IVB. (Reproduced from Ref. 6)
and TR216-.4, which were 0418 resistant but did not express the
216 gene-encoded antigen were analyzed further. Cells of these four
clones were injected into nude mice and fragments of the growing
tumors were used to challenge normal animals. The use of tumor fragments
grown in nude mice ensured that the cloned transfectants were still
capable of growing as a malignant tumor in nude mice. Table 1 shows
that the 216 gene expressing TR216 + .1 clone grew out in only one
of five animals and the 216 gene expressing TR216 + .2 clone was
rejected in all animals despite the fact that the mice were challenged
with a large tumor dose ( > 10 high 8 cells). In contrast, the clones
TR216- .3 and TR216-.4 which do not express the 216 gene-encoded
antigen grew in five of five and four of five mice, respectively,
and all these mice died of progressive tumor growth. The single
tumor which grew out in one of the animals that were challenged
with the 216 gene expressing TR216 + .1 clone was readapted to culture
and analyzed for the expression of the 216 gene-encoded antigen
with the fluorescence-activated cell sorter. All cells of the reisolate
were negative for the 216 antigen, indicating that the cells either
lost the transfected 216 gene or that its expression was prevented
by some other mechanism. This variant that did not express the 216
gene-encoded antigen was injected into five normal animals and tumor
growth resulted in all the mice (Table 1). Together, these data
indicate that the stable expression of the 216 gene-encoded antigen
is sufficient to change the phenotype of a progressor tumor so that
it is rejected by the normal animal. Furthermore, the loss of the
expression of this 216 antigen in transfected tumor cells allows
these cells to regain the progressor phenotype characteristic of
the untransfected parental progressor tumor.
C. Discussion
Many years ago, studies clearly demonstrated that tumor-specific
antigens that are distinct (unique) for each individual tumor can
lead to a complete immunological destruction of experimental cancers.
However, the molecules eliciting (and being the target of) these
immune responses have remained obscure. We have cloned and analyzed
the genes encoding novel class I molecules expressed by a UV -induced
murine skin tumor, designated 1591, to determine their role in the
immunobiology of tumor rejection and tumor progression. Several
lines of evidence clearly indicate that one of these genes, called
gene 216, encodes an antigen that elicits 1591 tumor-specific rejection
and is the target molecule of tumor rejection:
1. The 216 gene-encoded antigen must be lost before the tumor can
grow progressively in a normal immunocompetent mouse. Southern blot
analysis showed that all of the in vivo- or in vitro-derived progressor
variants analyzed had lost the 216 gene [6].
2. The molecule encoded by the 216 gene is specifically recognized
by the A antigcnspecific cytolytic T -cell clone that we have previously
shown to select in vitro for progress or variants from the parental
regressor tumor cell line.
3. The most conclusive evidence comes from the fact that transfection
of the 216 gene into progressively growing 1591 tumor variants leads
to the expression of the 216 gene-encoded antigen on the tumor and
to complete rejection of all cells expressing this antigen. Thus,
the progressor tumor reverted to the parental regressor phenotype
following transfection.
Unique tumor-specific transplantation antigens are antigenically
distinct for independently induced tumors. These different antigens
may, therefore, be encoded either by numerous different unrelated
genes or by a single gene which underwent multiple different mutational
changes. Alternatively, these antigens might be encoded by the members
of a gene family such as the immunoglobulin genes, the T -cell receptor
genes, the MHC class I and class II genes, or the genes of the multiple
retroviral proviruses which are present in the murine genome. Some
of these gene families are known to contain the coding information
for a large variety of distinct molecules and could therefore account
for the observed remarkable antigenic polymorphism among tumorspecific
transplantation antigens. It is interesting to notice that even
a single malignant cell can express multiple unique tumor-specific
antigens as has been shown for the tumor P815 [12] or 1591-RE [9].
To determine whether these antigens are encoded by a family of related
genes or by multiple unrelated genes, it is necessary to analyze
more tumors and to identify molecularly and genetically more unique
tumor-specific transplantation antigens. Another important and still
unresolved question regarding the origin of unique tumor-specific
antigens is whether the genes encoding such antigens are preexisting
in the genome or whether these genes appear as the result of somatic
mutation and as such re present the product of the mutagenic action
of carcinogens. Prcvious studies demonstrating unique antigenicity
of each of the independent transformants which were all derived
from one single parental cell seemed to suggest somatic carcinogen-induced
mutations as a likely mechanism [13]. However, it was not excluded
by these studies that the carcinogen treatment activated heritably,
but at random, different preexisting, previously silent genes. Such
a mechanism could also account for the observed immunogenicity of
tumors in the autochthonous host [2]. In order to determine whether
somatic mutations are involved in the malignant transformation and
in the generation of tumor-specific antigens, we are presently searching
for genetic changes in tumor cells which are not present in normal
cells of the same individual.
References
1. Prehn RT, Main JM (1957) Immunity to methylcholanthrene-indueed
sarcomas. JNCI 18:769
2. Klein G, Sjogren HO, Klein E, Hellstrom KE (1960) Demonstration
of resistance against methylcholanthrene-induced sarcomas in the
primary autochthonous host. Cancer Res 20.1561
3. Old LJ (1982) Cancer immunology. the search for specificity.
Natl Cancer Inst Monogr 60.193
4. Davey GC, Currie GA, Alexander P (1979) A serologically detected
tumor-specific membrane antigen of murine lymphomas which is not
the target for syngcneic graft rejection. Br J Cancer 40.168
5. Rouse BT, Roellinghoff M, Warner NL (1972) Anti-O serum-induced
suppression of cellular transfer of tumor-specific immunity to a
syngcncic plasma cell tumor. Nature (New BioI) 238.116
6. Stauss HJ, Van Waes C, Fink MA, Starr B, Schreiber H (1986) Identification
of a unique tumor antigen as rejection antigen by molecular cloning
and gene transfer. J Exp Med 164.1516
7. Stauss 111 , Linsk R, Fischer A, Banasiak D, Haberman A, Clark
I, Forman J, McMillan M, Schreiber H, Goodenow RS (1986) Isolation
of the MHC genes encoding the tumorspecific class I antigens expressed
on a murine fibrosarcoma. J Immunogenct 13:101-111
8. Philipps C, McMillan M, Flood PM, Murphy OB, Forman J, Lancki
O, Womack JE, Goodenow RS, Schreiber H (1985) Identification of
a unique tumor-specific antigen as a novel class I major histocompatibility
molecule. Proc Natl Acad Sci USA 82.5140
9. Wortzel RO, Philipps C, Schreiber H (1983) Multiple tumor-specific
antigens expressed on a single tumor cell Nature (Lond) 304.165
10. Wortzel RO, Urban JL, Schreiber H (1984) Malignant growth in
the normal host after variant selection in vitro with cytolytic
T cell lines. Proc Natl Acad Sci USA 81.2186
11. Philipps C, Stauss HJ, Wortzel R O, Schreiber I 1(1986) A novel
MI IC class I molecule as tumor-specific antigen. correlation between
the antibody-defined and the CTL-defined target structure. J Immunogenet
13.93
12. Uyttenhove C, Maryanski J, Boon T (1983) Escape of mouse mastocytoma
P815 after nearly complete rejection is due to antigenloss variants
rather than immunosupprcssion. J Exp Med 157.1040
13. Embleton MJ, Heidelberger C (1972) Antigenicity of clones of
mouse prostate cells transformed in vitro. Int J Cancer 9:8
|