|
* This work was supported by the MRC (UK). 1 Laboratory
of Gene Structure and Ex pression,
National Institute for Medical Research, The Ridgeway, Mill Hill,
London NW7 lAA, UK.
Introduction
The human ▀-globin gene cluster spans a region of70 kilobases (kb)
containing five developmentally regulated genes in the order 5'-epsilon,YGY
A ,delta,▀-3' (Fig. 1 ). The haematopoietic tissue in the early
stages of human development is the embryonic yolk sac and the epsilon-globin
gene is expressed. This is switched to the y-globin genes in the
foetal liver and the delta- and ▀-globin genes in adult bone marrow
(Fig. 2; for review, see [9]). High levels of these genes are expressed
in circulating red blood cells (RBC), giving rise to 90 % of the
total soluble protein. RBC are derived from a pluripotent stem cell
which can differentiate along alternate pathways to erythrocytes,
platelets, granulocytes, macrophages and lymphocytes. During the
transition to erythroblasts which have lost the capacity to proliferate,
the ▀-globin genes become transcriptionally activated achieving
messenger RNA (mRNA) levels of more than 25000 copies per cell.
A large number of structural defects have been documented in the
▀-globin gene locus (for review, see [9,44]). These defects are
responsible for a heterogeneous group of genetic diseases collectively
known as the ▀-thalassaemias, which are classified into ▀, delta
▀, y delta ▀, etc. thalassaemia subgroups according to the type
of gene affected. In a related condition, hereditary persistence
of foetal haemoglobin (HPFH), Y-globin gene expression and hence
HbF (fetal haemoglobin) production persist into adult life. These
clinically important diseases provide natural models for the study
of the regulation of globin gene regulation during development.
Most interesting in terms of transcription are the promoter mutations
and deletions. The delta ▀ thalassaemias and a number of the HPFHs
are associated with an elevated expression of the y-genes in adult
life as a result of deletions of varying size. Analysis of these
deletions has suggested that they act over considerable distances,
to influence differential gene expression within the human ▀-globin
domain.
The Locus Control Region
The existance of a region that activates the entire ▀-globin gene
cluster first became apparent from the study of a heterozygous Y-▀-thalassaemia
(Fig. 1 ) [31]. This patient contained one deletion allele which
lacked lOO kb, eliminating the entire upstream region but not the
▀-globin gene [57], which was shown to be completely normal [31,
64]. The other allele was expressed in the patient and it was therefore
not a lack of trans-acting factors which silenced the mutant chromosome
but an important control region had be missing. A set of developmentally
stable, hypersensitive sites, 5' HS 1, 2, 3 and 4, were shown to
be present upstream in the deleted region, and these
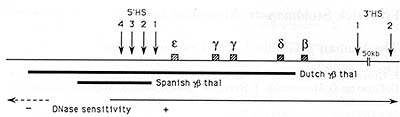
Fig. 1. Schematic representation of the ▀-globin locus.
Boxes indicate the different genes which are all transcribed from
left to right. The vertical arrows indicate DNase hypersensitive
sites. Thefour arrows mark the LCR containing 5' HS 1, 2, 3 and
4 upstream of the epsilon-gene.
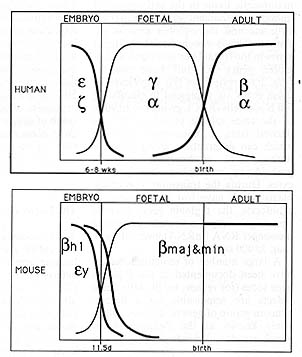
Fig. 2. Schematic representation of the developmental
expression patterns of globin genes in human and mouse. The curves
refer to the ▀-like genes only
were potential candidates for such a locus control region (LCR;
Fig. 1) [17,27 ,60]. Linkage of this region to a cloned ▀-globin
gene resulted in erythroid-specific high-level expression of the
gene in transgenic mice and in tissue culture cells. This expression
is dependent on the copy number of the transgene and independent
of the integration site in the host genome [3, 27], a phenomenon
which had not previously been observed in transgene expression.
This posed two questions: How is independence of the site of integration
achieved? And how is the LCR involved in globin gene switching?
Position independence and copy number dependence can theoretically
be explained by at least two independent mechanisms; either positive
activation by the LCR is always achieved, overriding position effects
that could be present, or (and) the region contains a locus border
element(s) (LEE) that insulate it from neighbouring regions. Matrix
attachment sites (MAR) [22, 30] or " A " elements [4, 53] could
be LEEs, and we initially speculated these were part of the LCR
in addition to activating sequences [27]. However, preliminary experiments
indicate that this is not the case and that such a border may be
located further upstream. The latter is based on the fact that the
DN aseI sensitivity of chromatin is strongly decreased in the sequences
25 30 kb 5' to the LCR (Fig.1) [19, 31, 57]. At least 150 kb of
chromatin in the 3' direction is sensitive under the control of
the LCR [19], suggesting that such sequences are not present for
a considerable distance 3' (Fig.1). The position independence we
observe is therefore due to a dominant activation of transcription
by the LCR, perhaps by creating very stable interactions between
the LCR and the genes. Consequently, positive position effects would
only be present in the background and only become apparent in situations
where the linked gene is suppressed [12] (see below for discussion).
Position effects are not observed at low levels of expression when
part of the LCR or mutations in the LCR are used. This indicates
that the interaction between the LCR and the promoter is dominant
except when the promoter is suppressed [10, 12, 18, 20, 49, 54].
In agreement with the deletion observed in a Hispanic y ▀-thalassaemia
(Fig. 1) [13], the main activity of the LCR is associated with HS2,
3 and 4 [10, 18, 20, 54, 61], which can each activate a linked transgene,
independent of the site of integration. A number of protein binding
sites have been mapped to these fragments, in particular, sites
for two erythroid-specific factors and several ubiquitously expressed
factors. A number of the binding sites are present in all three
active sites (Fig. 3) [43, 45, 55]. One of the shared factors is
the erythroid/megakaryocyte-specific factor GATA-1 [38,48], which
has been shown to be essential for erythroid development [42]. Deletion
of GA T A-1 binding sites prevents erythroid-specific induction
of the ▀-glo bin gene [ 11 ], and the protein has been shown to
have transcriptional activation properties [38]. However, the presence
of GATA-1 binding sites per se is insufficient to give position-independent
expression, e.g. the flanking regions of the human ▀-globin gene
contain at least six GATA-1 binding sites [11,63], but do not confer
integration site-independent expression [32,34,58]. However, all
three active 5' HS contain two closely spaced GA T A-1 sites in
opposite orientations. This arrangement is also observed in the
chicken ▀-globin enhancer, which appears to provide position-independent
expression [47]. Possibly an inverted double GATA-1 site is a key
component in erythroid -specific, posi tion -independent activation
and GA T A-1 can interact with itself or one of the other GA T A
proteins to achieve this [ 66] .Classical enhancer activity is only
associated with 5'HS2 [41, 61], and not with the others. Dissection
of the HS 2 showed that a number of proteins are bound to the core
fragment (Fig. 3) [55]. Attention has been focused on a double consensus
sequence for the jun/fos family of DNA binding proteins which appeared
crucial for HS 2 activity [41, 52, 55]. Several pieces of evidence
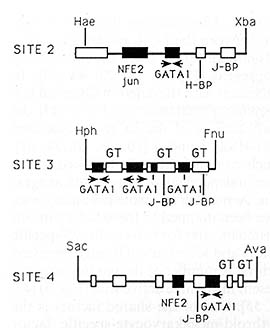
Fig.3. Summary of factor binding sites to the minimal
fragment of 5'HS2, 3 and 4, which provide position-independent expression
in transgenic mice. Individual factors are described in the text.
Black boxes indicate erythroid-specific factors; open boxes indicate
ubiquitous factors. GT indicates a GT -rich motif [43]
show that the functional activator which interacts with the jun/fos
binding site is NF-E2, originally described as interacting with
another erythroid-specific gene, that for porphobilinogen deaminase
[39, 40]. Two NF-E2 molecules and at least one other protein binding
at two nonequivalent sites are involved [56]. However, the presence
of this double NF-E2 sequence alone is insufficient to provide high
levels of expression [55] and when the jun/fos binding site is removed
from the 300 bp core fragment, HS2 retains the ability to activate
a linked ▀-globin gene in a copy number dependent fashion, albeit
at low levels (Fig. 4) [56]. We therefore conclude that the 5'HS2
NF-E2 region has strong enhancer activity but that it is not necessarily
required to obtain position-independent globin gene activation.
All the other factors which have been shown to interact with LCR
sequences, including the factors H-BP and J-BP, are ubiquitous proteins
[56]. This suggests that a combination of erythroid-specific and
ubiquitous factors may be required to render the ▀-globin gene independent
of its site of integration. The (abundant) ubiquitous factors shared
by the three HS of the LCR which have been studied to date are Sp
1 and TEF -2 [23, 65], but a simple multimerized combination of
a GATA-l and a Spl/TEF-2 binding site is not functional (S. Philipsen,
unpublished results). We therefore think that other, as yet less
well characterised factors may be involved in LCR function.
The LCR and Disease
The discovery, characterization and mapping of the LCR has enabled
the pursuit of two novel approaches to the study of globin-related
diseases. Firstly it allows high-Ievel expression of disease genes
such as the ▀ gene which is responsible for sickle cell disease.
By linking this gene in combination with human alfaglobin genes
several laboratories have succeeded in producing transgenic mice
which show sickle cell disease [26, 50, 59]. High levels of human
haemoglobin Scan be obtained in mice and the RBC of these mice show
a pronounced change in shape when deoxygenated (Fig. 5). We are
presently improving this model for two reasons, firstly, to study
the effects of sickle cell disease on the progression of infection
by different malaria strains, and secondly, to be able to study
the progression of sickle cell disease and the treatment thereof
by new protocols, in particular the development of gene therapy.
The latter has been given new hope by the mapping of the minimal
elements that give the full activity of the LCR. The LCR can now
be incorporated into retroviral vectors to develop therapy protocols
and preliminary experiments (F. Meyer, personal communication) indicate
that the LCR will provide high levels of expression in this context
in mice.
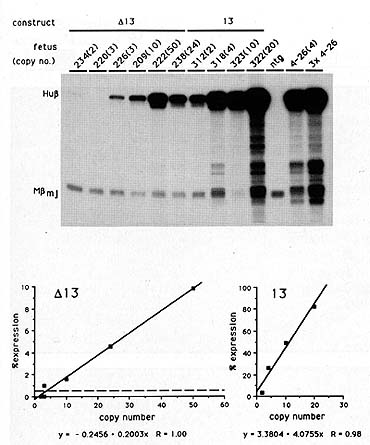
Fig. 4. S 1 nuclease analysis of HS 2 constructs containing
the NF -E2 sites (13) or not (1113) in transgenic mice. Foetal liver
RNA (day 13.5) was assayed using a mixed probe S 1 nuclease experiment
using the 5' human ▀-globin probe and the mouse ▀ maj probe [56].
Specific activities were 10: 1 for H u ▀ : M ▀ .Protected products
are indicated on the left. The 200 series of transgenic mice contains
the 1113 construct and the 300 series contains the 13 construct.
Copy numbers are shown in parentheses. Lower pnael depicts a quantitation
experiment of the S 1 protection analysis using the Hu▀5' probe
and a mouse alfa-globin probe as an internal control. The % expression
is given as the total Hu ▀-globin signal divided by the total mouse
alfa-globin signal (adjusted for specific activities). This was
plotted against the copy number. The line represents the result
of a linear regression analysis on the data points. The R value,
the correlation coefficient, indicates very high correlation with
a straight line (R = 1 ). The dashed line in the 1113 graph represents
the minimallevel that can be measured accurately
Developmental Regulation of the ▀-Globin Locus
Genetics.
The study of globin gene switching has been assisted by the characterization
of deletions and point mutations which affect expression of the
y and ▀-genes. Point mutations in the ypromoters have been linked
to HPFH phenotypes and these can be divided into two groups (Fig.
6). Those clustered around the distal CAA T box appear to result
in the loss of factor binding sites [21, 36], suggesting that this
region may contain a binding site for a negative regulator. For
example, a 13-bp deletion which removes the distal CAA T box results
in a very strong HPFH (60%) [25]. Interestingly, a recently described
Japanese HPFH (20%) is associated with a point mutation in the CAA
T sequence of the distal CAA T box
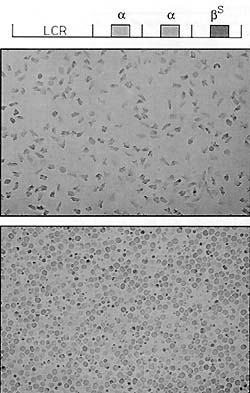
Fig. 5. Sickle cell disease in transgenic mice. The top
line shows the arrangement of genes and the LCR that was injected
into fertilized mouse eggs to obtain transgenic mice. The top panel
shows sickled cells from one of the transgenic mice [26]; the bottom
panel shows control nontransgenic red blood cells
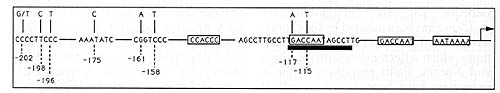
Fig. 6. Summary of mutations occurring in the y-globin
promoter resulting in HPFH phenotypes (see [44])
[21] which reduces affinity for the transcription factor CP 1. The
-117 mutation associated with Greek HPFH ( 40% ) has been reported
to cause reduced binding of the erythroid-specific factor NF-E3
[36]. These findings suggest a model for y silencing in which factors
binding to the distal CAA T box (at -115) compete for interaction
with factors bound to upstream promoter sequences preventing the
proximal CAA T box (at -87) from forming such interactions. The
distal CAA T box is located outside the normal optimal position
for CAA T elements, and this is likely to prevent it from functioning
as an effective positive promoter element. One would expect this
type of silencing mechanism to depend on the topology of the promoter
region and it is also likely to be affected by the creation of extra
factor binding sites in the upstream sequences. Such sites may partially
bypass the competition between the proximal and distal CAA T boxes,
resulting in suboptimal transcription. Indeed, a second group of
mutations, upstream of -150, result in new or improved binding sites
for transcription factors, e.g. Sp1 [16,28] and GATA1 [35, 37].
Activation of y transcription in the nondeletion HPFHs is associated
with down regulation ofthe▀-gene. The reduction in ▀ expression
(to around 60% in Southern Italian HPFH) is approximately equivalent
to the rise in expression of the cis-linked y-globin gene, with
only a slight reduction in overall transcriptional output from the
locus [24, 62]. This suggests that competition is taking place between
the genes and that this is tightly linked to the process of transcription.
However, a drastic reduction or loss of ▀ transcription due to point
mutations and deletions in the ▀-promoter does not significantly
increase y expression (less than 5%; Fig.7) [44]. Clearly, a y-gene
exerts a negative effect on the ▀-gene (coupled to transcription)
but this effect is not reciprocal. Some ▀-gene deletions show higher
levels of y expression (Fig. 7) but these deletions are always large
(> 10 kb). Of these, the Ay delta ▀ thalassaemias all have deletions
which extend into the region of y transcription. They are uninformative
for competition models because enhancers found near the deletion
breakpoints may be responsible for the high level, pancellular y
expression observed in the deletion HPFHs [1, 15]. Some increased
y expression is also observed in the delta ▀- and Dutch ▀ thalassaemias,
but the broad range of values between patients with the same deletion
and the heterocellular distribution of y-protein among the red cells
suggest that the increase in y expression is not solely at the transcriptional
level. This is supported by the observation that nontranscriptional
defects (e. g. RNA processing) in heterozygous ▀-thalassaemias cause
elevated levels of y chains (up to 5 %). Selection of a small proportion
of cells expressing y is a likely mechanism for this increase. Deletion
of the delta-gene (which is normally expressed at only 2% 3% of
the level of ▀) also does not seem to be required for the y-expression
observed in the delta ▀-thalassaemias, since it is intact in Dutch
▀-thalassaemia, which has a very similar phenotype. Instead, the
requirement appears to be a minimum size of deletion (> 10 kb).
Probably these large deletions perturb the chromatin structure of
the locus, resulting in a small increase in y transcription which
is further amplified by the chain imbalance. In conclusion, the
genetic data show that strong downregulation of the ▀-gene can result
from an increase of y-gene transcription, while there does not seem
to be any significant link between transcription of the ▀-gene and
silencing of the y-genes in adult life.
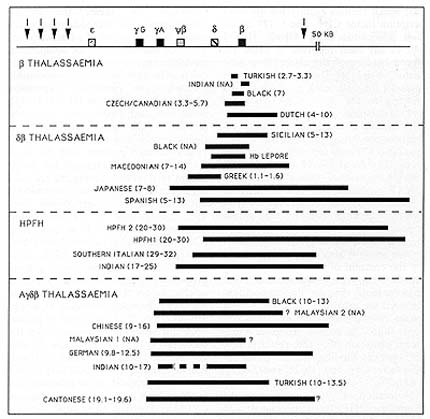
Fig. 7. Schematic representation of the different deletions
occurring in the ▀-globin locus in thalassaemias and HPFHs. Black
bars indicate the size of the deletion; numbers in parentheses indicate
the levels of y-globin expression in heterozygotes
Transgenic Mice.
Attempts to study switching of globin genes have also made use of
transgenic mice as a model system. Mice do not possess separate
foetal globin genes but instead switch directly from embryonic to
adult ▀-globin expression at 11-13 days of gestation (Fig. 2).
The developmental regulation of the human epsilon-gene has been
analysed in both embryonic stem cells and transgenic mice. In mice
the epsilon-gene is expressed at high levels during the embryonic
stage only when linked to the LCR and is completely silenced thereafter
[33, 46, 51]. Based on the studies by Cao et at. deletion mutants
lacking the -200 to -300 promoter region show a small increase in
epsilon expression in adult transgenic mice but the low level indicates
that other sequences may also be involved in silencing epsilon (P.
Fraser, unpublished observations). The human y-transgene without
the LCR is expressed like the mouse embryonic genes [7, 32]. It
was initially reported that linkage to the LCR resulted in y expression
at all developmental stages and that the y-gene was silenced in
adult mice only when the ▀-gene was also present. This appeared
to support a competition model where the ▀-gene is required for
silencing of the y-gene [2, 14]. However, a different result was
obtained when the single y-gene experiments were carried out on
animals carrying only one or two copies of the LCR-y-gene con
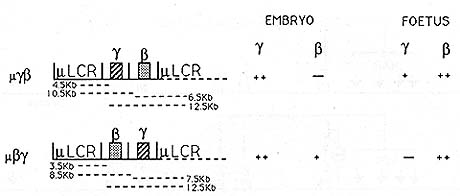
Fig.8. Microlocus (µLCR) y ▀ and ▀ y constructs
[29]. Genes are represented as shaded boxes. All genes are in the
same trancriptional orientation, 5' to 3', with respect to each
other and the LCR. The dotted LCR lines indicate the situation in
multicopy animals to illustrate the distance from a promoter to
a 5' and 3' LCR. These distances are indicated by dotted lines below
the constructs. Plus and minus symbols indicate high, medium, and
very low levels of expression
struct. y expression persisted in the early foetal liver, but was
silenced at adult stages, independent of the presence of the ▀-gene
[12]. Transcription of the LCRlinked y-gene can therefore also be
blocked completely by stage-specific negative regulators acting
on the sequences immediately flanking the gene, and this removes
the basis of the argument that the ▀-gene would be needed for y
silencing. The elements responsible for y silencing have not yet
been identified but the mutations associated with the nondeletion
HPFHs suggest that at least the sequences around the distal CAA
T box are likely to be involved (see above). The availability of
a transgenic mouse model for y-gene silencing should allow this
to be tested and possibly lead to novel approaches for treating
thalassaemia and sickle cell anaemia. If y-gene expression were
understood at the level of the transcription factors, it might be
possible to develop novel therapies that could specifically interfere
with the adult suppression of the y-gene and alleviate the clinical
problems associated with severe chain imbalance or sickling. Linkage
of the adult ▀-gene to the LCR results in inappropriate expression
at the embryonic stage [2,3, 14,29,33], albeit at a low level. Placing
a y-gene or a human alfa globin gene between the ▀-gene and the
LCR blocks this expression [2, 14, 29], supporting the idea that
competition plays a role in preventing premature ▀ expression. However,
when the order is reversed and the ▀-gene is placed in the first
position, it is expressed at a level similar to that observed for
the ▀-gene in the absence of the y- or alfa-gene (Fig. 8) [29].
Silencing of the ▀-gene at the embryonic stage is therefore not
caused by reciprocal competition only, but relative distance between
the LCR and the genes (i.e. position and polarity) is also important.
Polarity in the locus has long been suggested by the fact that the
genes are arranged in the order of their expression during development.
The order of the genes is conserved among mammals but there is some
divergence in the other vertebrate loci. In chicken, the embryonic
epsilon- and delta-genes are located at opposite ends of the locus,
with the adult ▀-genes between them. However, it is important to
note that the chicken ▀-globin LCR may have been split as part of
an epsilon translocation such that part of it is located between
the ▀- and epsilon-genes [8,47] and that the epsilon-gene contributes
only 20 % of the total embryonic haemoglobin compared to 80% for
delta [5]. The data reviewed above indicate that developmental regulation
of the human
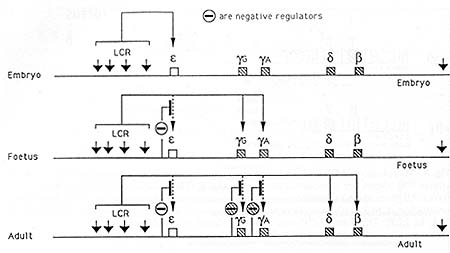
Fig. 9. Model for stage-specific regulation of the genes
of the ▀-globin locus. Solid lines indicate activation of genes
by the LCR. The symbol O indicates stage-specific negative factors
silencing the gene. The location of these is not accurate and there
may be more than one factor for each gene
▀-globin locus is a complex process which centres around developmentally
specific suppressors and the polarity of the locus (Figs. 9, 10).
The earliest gene to be activated, the epsilon-gene, is also the
one closest to the LCR. The y- and ▀-genes may be suppressed by
competition with epsilon; alternatively, or in addition, the y-
and ▀-genes may bind embryonic stage-specific factors which keep
their promoters suppressed. The epsilon-promoter is silenced in
the foetal liver by one or more suppressor factors, negating its
competitive ability (Fig. 9). As a result the y-genes are expressed,
and they in turn keep expression of the ▀-gene suppressed by competition.
The y-genes are switched off during the period around birth, again
by stagespecific negative regulators, and as a consequence the ▀-gene
is activated and expressed in the bone marrow. We propose that loop
formation between regulatory elements is the crucial parameter to
explain the suppression of the late genes by the early genes at
early stages but not vice versa. The frequency of interaction between
the promoters and the LCR will depend on the effective volume in
which these elements operate. This effect would be most pronounced
if the LCR and the genes were all present on one structural chromatin
loop several times the distance between the LCR and the genes. The
fact that the LCR controls DNase hypersensitivity of the ▀-globin
locus over at least 150-kb [19] suggests that the entire ▀ locus
may be present on one very large chromatin loop. If we assume that
to be the case, the frequency of interactions between any of the
promoters with the LCR would be proportional to their effective
concentration relative to the LCR (Fig. 10). On basis of ring closure
probabilities with naked DNA, the effective concentration of two
points on the DNA will be related to the volume of a sphere and
will be proportional to the power of3/2 of the distance. Applying
the rule to the ▀-locus, the ▀-gene is twice as far as the Gy-gene
from the HS 2 enhancer of the LCR. Therefore, the ▀-gene occupies
an approximately eight-fold larger volume relative to the HS-2 enhancer
of the LCR than the Gy-gene, which should give it a three-fold lower
frequency of interaction with the LCR (Fig. 10). This effect will
work in favour of the proximal gene, decreasing the affinity differences
required for competition, but it will work against the distal gene.
Distal genes would be incapable of suppressing upstream genes under
similar circumstances unless the downstream gene promoter increased
its affinity by several orders of magnitude relative to the upstream
gene. The transgenic mouse data on the expression of the ▀-globin
gene at the embryonic and foetal/adult stages argue strongly against
this possibility. Instead, the problem is solved by local suppression
of the upstream promoters to allow expression from the downstream
gene (Fig. 9). Experiments to substantiate or disprove this prediction
are presently in progress.
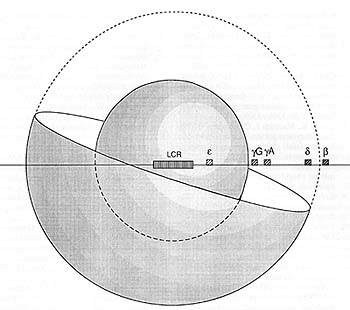
Fig.l0. Schematic representation of the relative volumes
occupied by the Gy- and ▀-genes relative to the LCR. For simplicity
of presentation the LCR is shown as a fixed point in the centre
of the sphere. Only half the ▀-globin gene outer sphere is shown
References
1. Anagnou NP, Perez-Stable C, Gelinas R, Costantini F, Liapaki
K, Constantopoulou M, Costeas T, Moschonas N, Stamatoyannopoulos
G (1990) ONA sequences residing 3' of the breakpoint of the HPFH-3
deletion can modify the developmental regulation of the fetal Ay
globin gene. Clin Res 38: 301 A
2. Behringer RR, Ryan TM, Palmiter RO, Brinster RL, Townes TM (1990)
Human y- to ▀-globin gene switching in transgenic mice. Genes Oev
4:380-389
3. Blom van Assendelft G, Hanscombe 0, Grosveld F, Greaves OR (1989)
The ▀-globin domain control region activates homologous and heterologous
promoters in a tissue-specific manner. Cell 56: 969-977
4. Bonifer C, Vidal M, Grosveld F, Sippel AE (1990) Tissue-specific
and position independent expression of the complete gene domain
for chicken lysozyme in transgenic mice. EMBO J 9: 2843- 2848
5. Brown J, Ingram V (1974) Structural Studies on Chick Embryonic
Hemoglobins. J BioI Chem 249:3960-3972
6. Cao S, Outman PD, Dave HPG, Schechter AJ (1989) Identification
of a transcriptional silencer in the 5'-flanking region of the human
epsilon-globin gene. Proc Natl Acad Sci USA 86: 5306-5309
7. Chada K, Magram J, Costantini F (1986) An embryonic pattern of
expression of a human fetal globin gene in transgenic mice. Nature
319: 685- 689
8. Choi O-R, Engel JD (1988) Developmental regulations of ▀-globin
gene switching. Cell 55: 17- 26
9. Collins FS, Weissman SM (1984) The molecular genetics of human
hemoglobin. Prog Acid Res Mol BioI 31:315-462
10. Collis P, Antonioti M, Grosveld F (1990) Definition of the minimal
requirements within the human ▀-globin gene and the dominant control
region for high level expression. EMBO J 9:233-240
11. de Boer E, Antonioti M, Mignotte V, Wall L, Grosveld F (1988)
The human ▀-globin promoter; nuclear protein factors and erythroid
specific induction of transcription. EMBO J 7:4203-4212
12. Dillon N, Grosveld F (1991) Human yglobin genes silenced independently
of other genes in the ▀-globin locus. Nature 350:252-254
13. Driscoll C, Dobkin C, Alter B (1989) Gamma/delta/beta thalassemia
due to a de novo mutation deleting the 5' ▀-globin gene locus activating
region hypersensitive sites. Proc Natl Acad Sci USA 86:7470-7474
14. Enver T, Raich N, Ebens AJ, Papayannopotiloti T, Costantini
F, Stamatoyannopotilos G (1990) Developmental regulation of human
fetal-to-adult globin gene switching in transgenic mice. Nature
344:309-313
15. Feingold E, Forget B (1989) The breakpoint of a large deletion
causing hereditary persistence of fetal hemoglobin occurs within
an erythroid DNA domain remote from the ▀ globin gene cluster. Blood
74:2178-2186
16. Fischer K, Nowock J (1990) The T to C substitution at -198 of
the Ay globin gene associated with the British form of HPFH generates
overlapping recognition sites for two DNA binding proteins. Nucleic
Acids Res 18:5685
17. Forrester W, Takegawa S, Papayannopotiloti T, Stamatoyannopoulos
G, Groudine M (1987) Evidence for a locus activation region: the
formation of developmentally stable hypersensitive sites in globin-expressing
hybrids. Nucleic Acids Res 15: 10159 -10177
18. Forrester W, Novak U, Gelinas R, Groudine M (1989) Molecular
analysis of the human ▀-globin locus activation region. Proc Natl
Acad Sci USA 86: 5439-5443
19. ForresterW, Epner E, Driscoll C, Enver T, Brice M, Papayannopotilou
T, Groudine M (1990) A deletion of the htiman▀ globin locus activation
region causes a major alteration in chromatin structure and replication
across the entire ▀ globin locus. Genes Dev 4:1637-1649
20. Fraser P, Hurst J, Collis P, Grosveld F (1990) DNasel hypersensitive
sites 1,2 and 3 of the human ▀-globin dominant control region directs
position-independent expression. Nucleic Acids Res 18: 3503-3508
21. Fticharoen S, Shimiza K, Ftiktimaki M (1990) A novel C- T transition
within the distal CCAA T motif of the Gy glo bin gene in the Japanese
HPFH: Implication of factor binding in elevated fetal globin expression.
Nucleic Acids Res 18: 5245
22. Gasser S, Laemmli U (1986) Cohabitation of scaffold binding
regions with upstream/ enhancer elements of three developmentally
regulated genes of D. melanogaster. Cell 46: 521- 530
23. Gidoni D, Kadonaga JT, Barrera-Saldana H, Takahashi K, Chambon
P, Tjian R ( 1985) Bidirectional SV 40 transcription mediated by
tandem Sp 1 binding interactions. Science 230: 511-514
24. Giglioni B, Casini C, Mantovani R, Merli S, Comp P, Ottolenghi
S, Saglio G, Camaschella C, Mazza U (1984) A molecular study of
a family with Greek hereditary persistence of fetal hemoglobin and
▀thalassaemia. EMBO J 11 :2641-2645
25. oilman J, Mishima N, Wen X, Stoming T, Lobel J, Huisman T (1988)
Distal CCAA T box deletion in the Ay globin gene of two black adolescents
with elevated fetal Ay globin. Nucleic Acids Res 18: 10635- 10642
26. Greaves DR, Fraser P, Vidal MA, Hedges MJ, Ropers D, Ltizzatto
L, Grosveld F (1990) A transgenic mouse model of sickle cell disorder.
Nature 343:183-185
27. Grosveld F, Blom van Assendelft G, Greaves D, Kollias G (1987)
Positionindependent high level expression of the human ▀-globin
gene in transgenic mice. Cell 51 :975-985
28. Gumicio D, Rood K, Gray T, Riordan M, Sartor C, Collins F (1988)
Nuclear proteins that bind the human y globin gene promoter: Alterations
in binding produced by point mutations associated with hereditary
persistence of fetal hemoglobin. Mol Cell BioI 8: 5310 -5322
29. Hanscombe 0, Whyatt D, Fraser P, Yannoutsos N, Greaves D, Grosveld
F (1991) Importance of globin gene order for correct developmental
expression. Genes Dev 5:1387-1394
30. Jarman A, Higgs D (1988) Nuclear scaffold attachment sites in
the human globin gene complexes. EMBO J 7:3337-3344 31. Kioussis
D, Vanin E, deLange T, Flavell RA, Grosveld F (1983) ▀-globin gene
inactivation by DNA translocation in Ythalassaemia. Nature 306:
662- 666
32. Kollias G, Wrighton N, Hurst J, Grosveld F (1986) Regulated
expression of human Ay-, ▀-, and hybrid y-▀-globin genes in transgenic
mice: manipulation of the developmental expression patterns. Cell
46:89-94
33. Lindenbaum M, Grosveld F (1990) An in vitro globin gene switching
model based on differentiated embryonic stem cells. Genes Dev 4:2075-2085
34. Magram J, Chada K, Costantini F (1985) Developmental regulation
of a cloned adult ▀-globin gene in transgenic mice. Nature 315:338-340
35. Mantovani R, Malgaretti N, Nicolis N, Ronchi A, Giglioni B,
Ottolenghi S (1988) The effects of HPFH mutations in the human y
globin promoter on binding of ubiquitous and erythroid specific
nuclear factors. Nucleic Acids Res 16:7783- 7797
36. Mantovani R, Superti-Fuga G, Gilman J, Ottolenghi S (1989) The
deletion of the distal CCAA T box region of the Ay globin gene in
black HPFH abolishes the binding of the erythroid specific protein
NFE 3 and of the CCAA T displacement protein. Nucleic Acids Res
17: 6681- 6691 37. Martin D, Tsai S, Orkin S (1989) Increased y
globin expression in anon deletion HPFH mediated by an erythroid-specific
DNA-binding factor. Nature 338:435438
38. Martin D, Orkin S (1990) Transcriptional activation and DNA
binding by the erythroid factor GF-1/NF-E1/Eryf 1. Genes Dev 4:
1886-1898
39. Mignotte V, Eleouet EF, Raich N, Romeo PH (1989) Cis- and transacting
elements involved in the regulation of the erythroid promoter of
the human porphobilinogen deaminase gene. Proc Natl Acad Sci USA
86:6548-6552
40. Mignotte V, Wall L, deBoer E, Grosveld F, Romeo P- H ( 1989)
Two tissue-specific factors bind the erythroid promoter of the human
porphobilinogen deaminase gene. Nucleic Acids Res 17: 37- 54
41. Ney PA, Sorrentino BP, Lowrey CH, Nienhuis A W (1990) Inducibility
of the HS II enhancer depends on binding of an erythroid specific
nuclear protein. Nucleic Acids Res 18:6011-6017
42. Pevny L, Simon MC, Robertson E, Klein WH, Tsai S, D' Agati V,
Orkin SH, Costantini F (1991) Erythroid differentiation in chimaeric
mice blocked by a targeted mutation in the gene for transcription
factor GATA-1. Nature 349:257-260
43. Philipsen S, Talbot D, Fraser P, Grosveld F (1990) The ▀-globin
dominant control region: hypersensitive site 2. EMBO J 9:2159-2167
44. Poncz M, Henthorn P, Stoeckert C, Surrey S (1989) Globin Gene
Expression in Hereditary Persistence of Fetal Hemoglobin and delta▀
Thalassaemia. Oxford University Press
45. Pruzina S, Hanscombe 0, Whyatt D, Grosveld F, Philipsen S (1991)
Hypersensitive site 4 of the human ▀-globin locus control region.
Nucleic Acids Res 19:1413-1419
46. Raich N, Enver T, Nakamoto B, Josephson B, Papayannopoulou T,
Stamatoyannopoulos G (1990) Autonomous developmental control of
human embryonic globin switching in transgenic mice. Science 250:
1147 -1149
47. Reitman M, Lee E, Westphal H, Felsenfeld G (1990) Site independent
expression of the chicken ▀A globin gene in transgenic mice. Nature
348:749- 752 48. Romeo PH, Prandini MH, Joulin V, Vignotte V, Prenant
W, Valnchenker W, Marguerie G, Uzan G (1990) Megakaryocytic and
erythrocytic lineages share specific transcription factors. Nature
334: 447-449
49. Ryan TM, Hehringer RR, Martin NC, Townes TM, Palmiter RD, Hrinster
RL (1989) A single erythroid specific DNaseI stiper-hypersensitive
site activates high levels of human ▀-globin gene expression in
transgenic mice. Genes Dev 3:314-323
50. Ryan T, Townes T, Reilly M, Asahura T, Palmiter R, Hrinster
R, Hehringer R (1990) A single erythroid specific DNaseI stiperhypersensitive
site activates high levels of human ▀-globin gene expression in
transgene mice.
51. Shih D, Wall R, Shapiro (1990) Developmentally regulated and
erythroid-specific expression of the human embryonic ▀globin gene
in transgenic mice. Nucleic Acids Res 18: 5465- 5472
52. Sorrentino HP, Ney PA, Hodine DM, Nienhaus A W (1990) A 46base
pair enhancer sequence within the locus activating regionis required
to induced expression of the y-globin gene during erythroid differentiation.
Nucleic Acids Res 18:2721-2731
53. StiefA, Winter DM, StratlingWH, Sippel AE (1989) A nuclear DNA
attachment element mediates elevated and positionindependent gene
activity. Nature 341:343-345
54. Talbot D, Collis P, Antonioti M, Vidal M, Grosveld F, Greaves
OR (1989) A dominant control region from the human ▀globin locus
conferring integration site independent gene expression. Nature
338:352-355
55. Talbot D, Philipsen S, Fraser P, Grosveld F (1990) Detailed
analysis of the site 3 region of the human ▀-globin dominant control
region. E MHO J9:2169-2177
56. Talbot D, Grosveld F (1991) The 5' HS2 of the globin locus control
region functions through the interaction of a multimeric complex
binding at two functionally distinct NF-E2 binding sites. E MHO
J 10:13911398
57. Taramelli R, Kioussis D, Vanin E, Hartram K, Groffen J, Hurst
J, Grosveld FG (1986) ydelta▀-thalassaemias 1 and 2 are the result
of a lOO kpb deletion in the human ▀- globin cluster .Nucleic Acids
Res 14:7017-7029
58. Townes T, Lingrel J, Chen H, Brinster R, Palmiter R (1985) Erythroid-specific
expression of human ▀-globin genes in transgenic mice. EMBO J 4:1715-1723
59. Trtidel M, Saadaen N, Gare M-C, Hardakdjian-Michau J, Blouquit
Y, Gtierquin-Kern J-L, Rotiyer-Fessard P, Vidatid D, Pachnis A,
Romeo P-H, Hetizard Y, Costantini F (1991) Towards a transgenic
mouse model of sickle cell disease: Hemoglobin SAD. E MHO J 10:3157-3165
60. Tuan D, Solomon W, Li Q, London I (1985) The "▀-like-globin"
gene domain in human erythroid cells. Proc Natl Acad Sci USA 82:6384-6388
61. Ttian D, Soloman W, London I, Lee DP (1989) An erythroid-specific
developmental-stage-independent enhancer far upstream of the human
"beta-like globin" genes. Proc Natl Acad Sci 86: 2554-2558
62. Weatherall DJ, Clegg JH (1981) The thalassaemia syndromes. Blackwell,
Oxford
63. Wall L, deHoer E, Grosveld F (1988) The human ▀-globin gene
3' enhancer contains multiple binding sites for an erythroid specific
induction of transcription. Genes Dev 2:1089-1100
64. Wright S, deHoer E, Rosenthal A, Flavell RA, Grosveld FG (1984)
DNA sequences required for regulated expression of the ▀globin genes
in murine erythroletikaemia cells. Phil Trans R Soc Lond B 307:
271 282
65. Xiao J, Davidson I, Macchi M, Rosales R, Vigneron M, Staub A,
Chambon P (1987) In vitro binding of several cell-specific and ubiquitous
nuclear proteins to the GT -I motif of the SV -40 enhancer. Genes
Dev 1:794-807
66. Yamamoto M, Ko L, Leonard M, Heug H, Orkin S, Engel J (1990)
Acitivity and tissue-specific expression of the transcrip tion factor
NF- E 1 multi gene family. Genes Dev 4: 1650 -1662
|