|
The Division of Biological Sciences and the Pritzker
School of Medicine, Section of Hematology/Oncology,
Box 420, 5841 S. Maryland Avenue, Chicago/lllinois 60637
A. Introduction
The close association of specific chromosome abnormalities with
particular types of human cancer has been established by a number
of investigators during the past decade [1-6]. A few of the genes
involved in consistent chromosome rearrangements, notably translocations,
have already been identified, and it is likely that the identity
of most of the genes affected by these aberrations will be determined
within the next decade. Moreover, for several of the rearrangements,
some of the changes in gene structure and function have been defined.
Therefore, some general principles that may be applicable to many
chromosome rearrangements in human malignant disease are beginning
to emerge. Chronic myeloid leukemia (CML) provides one of the clearest
examples of our progress in first identifying a recurring chromosome
abnormality and then cloning the genes involved in the abnormality.
The analysis of these genes and their alteration as a result of
the chromosome change is the subject of this lecture.
B. Cytogenetic and Clinical Features of Chronic Myeloid Leukemia
Chronic myeloid leukemia is important because it was the first
human cancer in which a consistent chromosome abnormality was identified.
The abnormality is the Philadelphia or Ph I chromosome [7], which
was shown with banding to involve No.22 (22q -). The correct chromosome
defect was shown to be a translocation involving Nos. 9 and 22;
this was the first consistent translocation specifically associated
with any human or animal disease (Fig. 1) [8]. The reciprocal nature
of the translocation was established only recently, when the Abelson
protooncogcnc, ABL. normal1y on No.9, was identified on the Ph1
chromosome [9]. Other studies with fluorescent markers or chromosome
polymorphisms have shown that, in a particular patient, the same
No.9 and No.22 are involved in each cell. The Ph 1 chromosome is
present in granulocytic, erythroid, and megakaryocytic cells, in
some B cel1s, and probably in a few T cells. The karyotypes of many
Ph 1+ patients with CML have been examined with banding techniques
by a number of investigators; in a review of 1129 Phi+ patients,
the 9;22 translocation was identified in 1036 (92% ) [4]. Variant
translocations have been discovered, however, in addition to the
typical t(9;22). Until very recently, these were thought to be of
two kinds; one appeared to be a simple translocation involving No.22
and some chromosome other than No.9 (about 4%), and the other was
a complex translocation involving three or more different chromosomes,
two of which were No.9 and No.22 (about 4%). Recent data clearly
demonstrate that No.9 is affected in the simple as wel1 as the complex
translocations, and that its involvement had been overlooked [10].
Virtual1y al1 chromosomes have been involved in these variant translocations,
but No.17 is affected

Fig. I. Trypsin-Giemsa-stained karyotype of a metaphase
cell from a bone marrow aspirate obtained from an untreated male
with CML illustrating the t(9;22) (q34;q11 ). The Philadelphia chromosome
(Ph 1) is the chromosome on the right in pair 22. The material missing
from the long arm of this chromosome (22q -) is translocated to
the long arm of chromosome 9 (9q +), and is the additional pale
band that is not present on the normal chromosome 9
more often than are other chromosomes. The genetic consequences
of the standard t(9;22) or the complex translocation involving at
least three chromosomes is to move the ABL protooncogene on No.9
next to a gene on No.22, called B CR, whose function is currently
unknown (Fig. 2). Chronic myeloid leukemia usually terminates in
an acute leukemia in which the blast cells have either lymphoid
or myeloid morphology. In the acute phase, about 10%-20% appear
to retain the 46, Ph 1+ cell line unchanged, whereas most patients
show additional chromosome abnormalities resulting in cells with
modal chromosome numbers of 47 to 50 [4]. Different abnormal chromosomes
occur singly or in combination in a distinctly non random pattern.
In patients who have on I y a single new chromosome change, this
most commonly involves a second Ph 1, an isochromosome for the long
arm of No.17 [i(17q)], or a +8, in descending order of frequency
.Chromosome loss occurs only rarely; that most often seen is -7,
which occurs in 3% of patients.Early cases of acute leukemia in
which the Ph 1 chromosome was present were classified as CML presenting
in blast transformation; at present, patients who have no prior
history suggestive of CML are classified as Ph 1+ acute leukemia.
In fact, some of the patients with Ph 1+ ALL have a different breakpoint
in the B CR gene on No.22. In blast crisis, some blasts have intracytoplasmic
IgM, which is characteristic of pre-B cells, and these cells have
an immunoglobulin gene rearrangement [11].
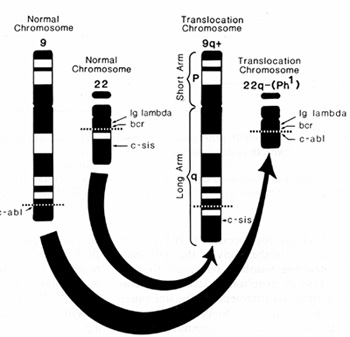
Fig. 2. Schematic drawing of chromo some No.9 and No.22
illustrating the chromosome translocation that produces the 9q +
and 22q- (Phi) chromosomes. One protooncogene. ABL, is moved to
No.22 adjacent to a gene of unknown function called BCR; the break
in No.22 is distal to the IG lambda locus which is not in volved
in the translocation. The SIS protooneogene is moved to the 9q +
chromosome. It is located at some distance from the breakpoint on
No 22 and there is no evidence that it is altered as the result
of the translocation
Marrow cells from some patients appear to lack a Ph 1 chromosome.
The majority of these patients had a normal karyotype. Somewhat
surprisingly, the survival of these patients was substantially shorter
than those whose cells were Ph 1 + [12]. Our recent review of the
histology of 25 Ph 1 patients showed that most of them did not have
CML but they had some type of myelodysplasia, most commonly chronic
myelomonocytic leukernia or refractory anemia with excess blasts
[13]. However, the situation has become more complex because it
has been shown recently that some patients with clinically typical
CML who lack a Ph 1 chromosome cytogenetically have evidence of
the insertion of A BL sequences into the BCR gene [14,15]. Thus,
it can be proposed that the sine qua non of CM L is the juxtaposition
of BC R and ABL.
C. Molecular Analysis of the 9;22 Translocation
Investigators are now in the process of unraveling the mystery
of the Ph 1 translocation in CML and ALL. In the t(9;22) in CML
and ALL, the Abelson protooncogene (ABL) is translocated to the
Ph 1 chromosome [9]. The ABL gene was first identified because of
its hornology to the viral oncogene that had been isolated from
a mouse pre-B-cell leukemia. The breakpoint junction in CML was
cloned and the site on the Ph 1 was called bcr, for breakpoint cluster
region, [16] since the majority of breaks cluster in a small 5.8-kilobase
(kb) region. The gene in which this cluster is located has also
been cloned. It is a very large gene greater than 100 kb and it
is presently also called BC R, which leads to a great deal of confusion.
In this lecture, bcr is used to denote the CML breakpoint region
and BCR to identify the whole gene. In contrast to bcr, the breaks
in ABL on No.9 occur over an incredible distance of more than 200
kb. We have used pulse
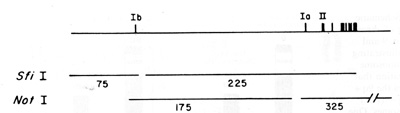
Fig.3. Map of the A BL gene showing the position of the
two alternative exons Ib and la relative to exon II Exon Ib is the
most 5' exon, whereas la is less than 20 kb from exon II which is
the common splice acceptor site. The vertical bars above the horizontal
line represent the more 3' exons which are homologous to the v-abl
sequences. S.11I and Not I are the enzymes used to determine the
relative positions of exons la, Ib, and II. (Figure adapted from
[17])
field gel electrophoresis (PFGE) to great advantage in the study
of the ABL protooncogene. Southern blotting with standard gel electrophoresis
leads to separation of DNA fragments in the size range of 2 to about
25 kb. Since the ABL gene is larger than 200 kb, mapping it in 10-
to 20-kb pieces is a formidable task. In contrast, by using PFGE
one can separate fragments more than 1000 kb in size, and this technique
is also very effective in the 100- to 600-kb range. Anormal chromosome
band contains roughly 500010000 kb and, thus, several very large,
overlapping fragments could contain a single band. Using many probes
for ABL provided by various investigators, Drs. Westbrook and Rubin
have constructed a map of the normal ABL gene [17]. This is a very
complex gene that normally uses one of two alternative beginnings.
exon Ia or Ib. During transcription, either of these can be spliced
at the same point on the remainder of the gene, which is called
the common splice acceptor site or exon II (Fig. 3). One of their
first discoveries was that the type Ib exon mapped more than 200
kb upstream from exon II. As a result, a very large segment of the
RNA transcript is removed or spliced out to form the mature mRNA.
This is a remarkable feat, not identified before in biological systems.
The breakpoints in the chromosomes of various CML patients and cell
lines occur in many locations upstream (5') of exon II. However,
the same size (8.5 kb) mRNA is found in all CML patients; this occurs
because the BCR exons arc spliced to ABL exon II, resulting in a
chimeric mRNA which is translated into a chimeric protein (p210IJCR-AIJL)
[18, 19]. With regard to Ph l-positive ALL, it has always been an
enigma why the typical Ph 1 translocation is seen in ALL and in
fact is the most common translocation in adults with ALL l20]. One
relatively trivial explanation would be that the patients really
had CML in lymphoid blast crisis with an undiagnosed chronic phase,
and this may occur in some patients. However, analysis of DNA from
some Ph l-positive ALL cells indicates that the breakpoint in No.22
is outside the bcr region. In one study, the majority of adult patients
(13 of 17) appeared to have the same bcr rearrangement that is seen
in CML whereas it has not been found in any or 7 children, who presumably
had a more 5' breakpoint in the BCR gene [21] (Table 1). Data from
our
Table 1. Ph 1-positive leukaemia
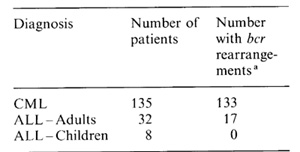
a bcr rearrangement in CML breakpoint
cluster region
laboratory as well as others indicate that the breakpoints on No.22
are greater than 50 kb proximal to the CML break but that they still
are within the BCR gene [22]. The breakpoints on No.9 are similar
to those in CML. Several investigators have shown that these Ph
I + ALL patients have an abnormal size chimeric BCR-ABL mRNA (7.0
7.4 kb) and ABL protein (p185BCR-ABI) [23,24]. These discoveries
and the development of DNA probes that can detect rearrangements
in the BC R and A BL genes have been applied very rapidly for use
in diagnosis and monitoring of patients thought to have CM L or
Ph 1+ ALL. The results of the diagnostic use of the bcr probe are
summarized in Table 1. Equally important is the ability to check
for the recurrence of a Ph 1 + cIone in CML patients who have undergone
bone marrow transplantation or in Ph 1 + ALL patients in remission.
These screening procedures have become even more sensitive with
the use of the poIymerase chain reaction to detect the bcr-ABL junction
in Ieukemic cells. We have recentIy studied seven patients with
Ph 1 + ALL to determine whether the transIocation breakpoints all
occur within the BC R gene [25]. With PFGE we could show that every
patient had a rearrangement within the BCR gene either in the 5'
portion of BC R in the first intron (five patients) or in bcr (two
patients). Moreover ABL was fused with BCR in each patient. Of the
seven patients, two were children, one of whom, age 12 years, had
a bcr rearrangement. Further studies with additionaI patients will
allow more precise correlations of the clinical features of the
Leukemias with the molecular abnormalities that underlie them. In
the future, we will understand the role of the BCR and ABL proteins
in normal cells and that of the two different chimeric BCR-ABL proteins
in CML and in ALL. Thus, the genetic analysis of what appeared to
be a simple chromosome change, namely the 9;22 translocation, has
revealed unexpected complexity. I am sure that, in the future, an
understanding of the altered function of the ABL protein will be
central to the development of more specific and more effective forms
of therapy.
D. BiologicaI Significance of Chromosomal Rearrangements
One of the most surprising revelations in the recent past has involved
the cellular oncogenes and their chromosome location (Fig.4). Much
of the excitement derives from the observation that many protooncogenes
are located in the bands that arc involved in consistent translocations
[3, 6]. There is a remarkable specificity of certain chromosome
rearrangements for particular subtypes of tumors especially leukemia
or lymphoma. The mechanism or mechanisms by which this specificity
is achieved are unknown; however, a number of investigators have
shown that certain proteins required for promotion of gene expression
are synthesized in a very cell-type-specific manner [26]. These
proteins are only present in the appropriate cell type and therefore
the particular gene is activated only in that cell type. The chromosome
rearrangements affecting M YC in B-cell [27, 28] and T -cell l29,
30] tumors strongly support the interpretation that the specificity
resides in the gene that is uniquely active in the particular cell
type. Thus the immunoglobulin genes are highly regulated in B cells
and they can therefore serve as the switch or activator mechanism
for MYC in B cells; on the other hand, the alpha chain of the T
-cell receptor (TCRA) is an active gene in T cells with a strong
enhancer/promotor and it clearly is an activator for M YC in T cells.
A reasonable paradigm is that translocations bring together, in
an inappropriate manner, a growth factor or growth factor receptor
gene (the protooncogene in the examples defined to date) adjacent
to an active cell-specific gene.
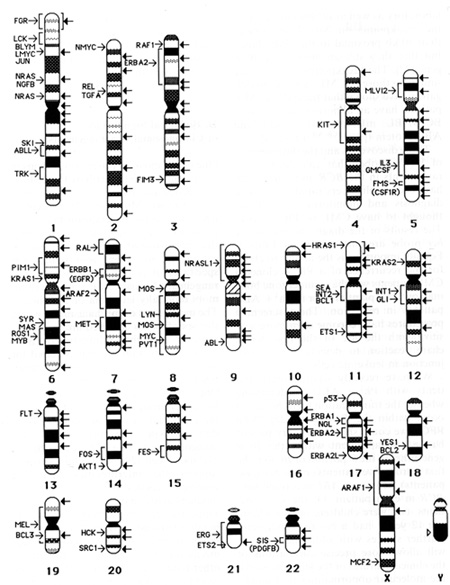
Fig.4. Map of the chromosome location of protooncogenes
or of genes with transforming properties and the breakpoints observed
in recurring chromosome abnormalities ill human leukemia, lymphoma,
and solid tumors. The protooncogenes and their locations are placed
to the left of the appropriate chromosome band (arrow) or region
(indicated by a bracket). The breakpoints ill recurring translocations,
inversions, deletions, etc., are indicated with all arrow to the
right of the affected chromosome band. The locations of the cancer
specific breakpoints are based on the Human Gene Mapping 9 report
[5]
It should be emphasized that many of the protooncogenes were identified
in viruses that cause tumors. However, these genes have not been
conserved through evolution from yeast and Drosophila to the chicken,
mouse, and man to cause cancer! Where we have any insight into the
function of these genes in normal cells, they are growth factors
or growth factor receptors. It is not unexpected that the genes
which a virus might coopt if it developed into a tumor-producing
virus would be genes that control prolifcration, genes which under
viral regulation would function abnormally with regard to cell growth.
Further support for the concept that oncogenes are growth factors
gone wrong is provided by studies at the Hall Institute in Melbourne.
There, investigators inserted the cloned gene for granulocyte-macrophage
colony-stimulating factor into a viral vector, transfected mouse
myeloid cells with this gene, and then injected the cells into mice
which developed leukemia [31]. The term "oncogene" is too short
and easy for it to be discarded, but it really refers to respectable
genes for growth factors or their receptors. The analysis of various
tumors for alterations in protooncogenes has revealed that a number
are abnormal as a result of translocations, amplification, or mutations
[32]. In some situations the relationship of the change in the protooncogene
to the multistage process of malignant transformation is unclear
[33]. Such ambiguity is not a problem with chromosome translocations;
the evidence is overwhelming that the t(8;14) in Burkitt's lymphoma
and the t(9;22) in CM L are an integral component of the cascade
of events leading to the transformation of a normal to a malignant
cell. The everincreasing numbcr of translocations reviewed in this
chapter provide a potential gold mine for identifying new genes
that are unequivocally related to the malignant phenotype of the
affected cell. The challenge is to isolate these translocation breakpoint
junctions, to identify the genes that are located at these break
points, and then to determine the change in gene function that occurs
as a consequence of the translocation. The ultimate measure of success,
however, will be in the application of these new insights in the
development of new, more effective treatments for cancer. In the
future, each particular subtype of tumor will be treated in a uniquely
defined way that is most appropriate for the specific genetic defect
present in that tumor. This should lead to a new era of cancer therapy
that is both more effective and less toxic.
References
I. Mitelman F ( 1988) Catalog of chromosome aberrations in cancer
Liss, New York
2. Heim S, Mitelman F (1987) Cancer cytogenetics. Liss, New York
3. Rowley JD (1988) Chromosome abnor malities in leukemia. J Clin
Oncol 6.194-202
4. Rowley JD, Testa JR (1983) Chromosome abnormalities in malignant
hematologic diseases In: Advances in cancer research, Academic,
New York, pp 103-148
5. Bloomfield CD, Trent JM, Van den Berghe I-I (1987) Report of
the eommittee on structural chromosome changes in neoplasia (HG
M9). Cytogenet Cell Genet 46: 344- 366
6. Yunis JJ (1983) The chromosomal basis of humanneoplasia. Science
221:227-236
7. Nowell PC, Hungerford DA (1960) A minute chromosome in human
granulo cytic leukemia Science 132.1497
8. Rowley JD (1973) A new consistent chro mosomal abnormality in
chronie myelo genous leukemia. Nature 243.290 -293
9. De Klein A, Van Kessel AG, Grosveld G et al. (1982) A cellular
oncogene is translocated to the Philadelphia chromosome in ehronic
myelocytie leukemia. Nature 300.765-767
10. De Klein A, Hagemeijer A (1984) Cytogenetie and molecular analysis
of the Ph 1 translocation in chronie myeloid leukemia. Caneer Surv
3.515-529
II. Bakhshi A, M inowada J, Arnold A et al. (1983) Lymphoid blast
erises of chronie myelogenous leukemia represent stages in the development
of B-eeIl precursors. N Engl J Med 309 826-831
12. Whang-Peng J, Canellos GP, Carbone PP ct al (196S) Clinical
implications of cytogenetic variants in chronic myclocytic Icukemia
(CM L). Blood 32: 755 766
13. Pugh WC, Pearson M, Vardiman JW ct al. (19S5) Philadclphia chromosomc-negative
chronic myclogenous leukaemia. a morphologic reassessment. Br J
Haematol 60457-467
14. Morris CM, Reeve AF, Fitzgerald PH ct al. (1986) Genomic diversity
correlates with clinical variation in Ph1-negativc chronic mycloid
leukemia. Nature 320. 281 2S3
15. Bartram CR ( 1988) Molecular genetic analyses of chronic myelocytic
Ieukemia. In Huhn D, Hellriegel KP, Nicdcrlc N (eds) Chronic myclocytic
leukemia and interferon. Springer, Berlin Heidelberg New York
16. Groffen J, Stcvcns()n J R, Heisterkamp N et al (1984) Philadelphia
chromosomal breakpoints are clustered within a limited region, bcr,
on chromosomce22. Cell 36. 93-99
17. Westbrook CA, Rubin CM, Carrino JJ ct al. (19SS) Long-range
mapping of the Philadelphia chromosome by pulsed-field gel electrophoresis
Blood 79.697-702
18. Konopka JB, Watanabe SM, Witte ON (1984) An altcration of the
human c-ahl protein in K562 Ieukcmia cells unmasks associatc tyr()sinc
kinase activity. Cell 37:1035-1042
19. Shtivelman E, Lifshitz B, Gale R P ct al (19S5) Fused transcript
of ahl and bcr genes in chronic myclogenous Ieukaemia. Nature 315550
554 20 Third international workshop on chromosomes in Ieukcmia (1982).
Cancer Genet Cytogenet 495-142
21. Dc Klein A, Hagemeijcr A, Bartram CR ct al (1986) Rearrangement
and translocation of the c-ahl oncogene in Philadclphia positivc
acutc Iymphoblastic lcukcmia Blood 681369-1375
22. Rubin CM, Carrino JJ, Dickler MN ct al ( 19SS) Heterogcncity
of genomic fusion of BC R and A HI in Philadclphia chromosome-positive
acute lymphoblastic Icukcmia Proc Natl Acad Sci USA S5.2795-2799
23. Clark SS, McLaughlin J, Christ WM et al. (19S7) Unique forms
of the ahl tyrosine kinase distinguish Ph I-positive CM L from PH
I-positive ALL. Science 235. S5 88
24. Chan LC, Karhi KK, Rayter SI et al ( 1987) A novel ahl protein
expressed in Philadelphia chromosome positive acutc Iymph()blastic
Icukacmia. Naturc 325. 635-637
25. Hooberman A, Carrino JJ, Leibowitz D et al. (1989) Unexpected
hetcrogencity of BcR-ABL fusion mRNA detected by polymerase chain
reaction in Philadclphia chromosome acute Iymphoblastic Ieukemia
Proc Natl Acad Sci USA S6.4259-4263
26. Nomiyama H, Fromental C, Xiao JH ct al. ( 19S7) Cell-specific
activity of the constituent elements of the simian virus 40 enhancer.
Proc Natl Acad Sci USA 84.7881 -7885
27. Leder P, Battey J, Lenoir G ct al. (1983) Translocations among
antibody genes in human cancer. Science 222: 765- 771
28. Croce CM, Isobe M, Palumbo A et al. ( 1985) GCl1C for x-chain
of humal1 T -cell receptor. location on chromosome 14 region involved
in T -cclll1coplasms. Scicl1ce 2271044-1047
29. Shima EA, Le Beau MM, McKeithal1 TW ct al (1986) Gene encoding
the alfa chain of the T -cell receptor is moved immediately down
stream of c-myc in a chromosomal 8;14 translocation in a cell line
from a human T -cell leukemia. Proc Natl Acad Sci USA 83.3439-3443
30. Mathieu-Mahul D, Caubet JF, Bernheim A ct al (1985) Molecular
cloning of a DNA fragment from human chromosome 14 ( 14q 11) involved
in T cell malignancics. EM 130 J 4 3427- 3433
31. Lang RA, Metcalf D, Gough NM et al. (1985) Exprcssion of a hemopoietic
growth factor cDNA in a factor-dependent cell line results in autonomous
growth and tumorigenicity. Cell 43.531- 542
32. Bishop J M ( 1987) The molecular genetics of cancer. Science
235. 305 -311
33. Duesberg PH (1987) Retroviruses as car cinogens and pathogens:
expectations and reality Cancer Res 47.1199-1220
|