|
Deutsches Krebsforschungszentrum, Im Neuenheimer Feld 280, 6900 Heidelberg, FRG Table 1 lists possible interactions of viruses in oncogenesis and emphasizes the role of human pathogenic viruses. The majority of tumor viruses known today insert their genetic material into the host cell nucleus, where it persists. The expression of at least one viral function appears to be a prerequisite for the maintenance of the transformed state. An interesting difference in interactions in oncogenesis is noted in infections by herpes simplex viruses. Abortive infection by these viruses modifies the host cell DNA [30, 31]. Transformation of rodent cells by these infections does not require persistence of viral DNA, but selective DNA amplification induced by these agents may playa crucial role in transformation [15]. Recently, it has been noted that besides herpes simplex viruses other members of the herpesvirus group, such as the pseudorabies virus (B. Matz, personal communication) and murine cytomegaloviruses (R. Heilbronn, unpublished data) share this property. Even a member of the poxvirus group, vaccinia, is able to induce selective DNA amplification [32]. It remains to be seen whether this property is consistently linked to transforming functions. A third group of agents is represented by viruses causing the acquired immunodeficiency syndrome (AIDS). In this condition, specific types of tumors develop as a consequence of the depressed immune function without being directly related to the AIDS virus infection. The fourth rather interesting group is represented by agents suppressing oncogenicity. This property has thus far been observed exclusively in infections with autonomous or helper-dependent parvoviruses. Their mode of interaction with infected host cells may provide some clues on early events in carcinogenesis. Available evidence suggests an interference of these viruses with early events, Table 1. Interactions of viruses
with hosts and hosts cells in oncogenesis  Table 2. Viruses involved in human oncogenesis  Table 3. Approximate latency periods for virus-associated human cancers 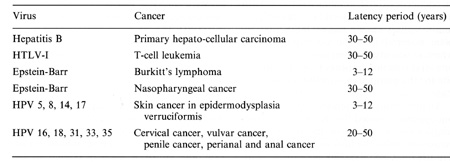 possibly related to initiation. in oncogenesis [15]. The following contribution concentrates on viruses in human tumors inserting their genetic material within the host nucleus. Members of four groups of viruses have thus far been identified as playing a role in human tumors. These viruses and the respective human tumors are listed in Table 2. The percentage of human tumors which can be linked to these infections on a worldwide scale is remarkably high, at present approximately 15% for both sexes [41]. In females close to 20% of all cancers can be linked to viral infections, most notably cervical cancer, amounting to almost 16%. In males slightly less than 10% are virally linked; 6.2% represent primary hepatocellular carcinomas. All cancers developing as a consequence of these viral infections share some characteristics: 1. They are monoclonal. 2. They develop only after long latency periods (in most instances after several decades; see Table 3). 3. Only a small percentage of infected individuals eventually develop cancer. 4. Expression of the persisting viral genome is the rule but appears to be restricted to a specific set of genes. Attempts to clarify the role of viruses in human tumors will have to take into account these observations. During the past few years a remarkable role of viruses of the papillomavirus group in human cancer has become evident. Particularly human anogenital cancer (including cervical, vulvar, penile, and perianal cancer) and specific rare forms of human skin cancer have been linked to these infections [43]. The mechanism of cancer induction following some of these infections may provide clues for the understanding ofvirus linked oncogenesis in man and will be discussed in more detail subsequently. Observations made by Rigoni-Stern (published in 1842) on a vastly different incidence of cervical cancer in prostitutes as compared with nuns mark the first important contribution to etiological factors in human genital cancer [27]. These studies, confirmed by many subsequent analyses, stimulated research on the role of sexually transmitted agents in the genesis of this neoplasia. Up to the end of the 1960s speculations were made concerning a possible role of gonorrhea, syphilis, and trichomoniasis in the induction of genital cancer, without there being any really supportive epidemiological evidence [28]. At the end of the 1960s a significant change occurred: two groups almost simultaneously reported seroepidemiological data implying an involvement of herpes simplex virus (HSV) type-2 infections in the etiology of cervical cancer [23, 26]. During the following 10 years a surprising number of confirmatory manuscripts were published, including experimental data on viral reactivation from tumor tissue, on the presence of a variety of HSV -specific antigens, on viral ribonucleic acid, and (much more scarce) on fragments of viral DNA within premalignant and malignant tissue (reviewed in zur Hausen [40]). Since partially inactivated HSV was also shown to induce transformation of rodent cells [9], by the end of the 1970s HSV appeared to be a strong candidate for a role in the induction of cervical cancer. In 1969 my group in Würzburg attempted to demonstrate Epstein-Barr virus DNA in Burkitt's lymphomas and nasopharyngeal cancer. After succeeding in the purification of EBV DNA, it was readily possible to find genomes of this herpes-group virus regularly in Burkitt's lymphomas and nasopharyngeal cancer biopsies by DNA-DNA or DNARNA hybridizations [42, 44]. Therefore, it was an obvious task to use the same technology in attempts to find HSV DNA in anogenital cancer. Somewhat unexpectedly, we encountered difficulties. The analysis of a large number of biopsies provided exclusively negative results [45]. In early 1972 I became convinced that HSV, if at all involved in genital cancer, could not interact with host cells analogous to other tumor virus systems known at that time by leaving persisting viral DNA with partial gene expression in the transformed cell. Since another mode of interaction appeared to be unlikely at this period, and cervical cancer nevertheless remained a strong candidate for a viral etiology, during 1972 we started looking for a role of other viruses in human genital cancer . There were good reasons for selecting human papillomavirus (HPV) for the subsequent study: a virus had been known since 1907 [4] to be the causative factor of human warts. Genital warts, a particularly unpleasant infection characterized by exuberant exophytic growth, had been shown electron microscopically to contain viral particles morphologically identical to those in common warts [1]. More importantly, there existed a substantial number of anecdotal reports, published over almost a whole century, on malignant conversion of genital warts (condylomata acuminata), usually after long duration and therapy resistance (reviewed in zur Hausen [37]). Wart viruses had barely been characterized at this period: this was mainly due to the lack of in vitro systems for viral propagation and to a remarkable host specificity of these viruses [29]. Studies depended entirely on the availability of biopsy materials containing large quantities of viral particles. Our initial studies were performed with cRNA preparations obtained from an individual plantar wart. The first hybridization data published in 1974 [45] showed that many, but by far not all, DNAs from common warts hybridized with the radioactive probe. N one of the genital warts and none of the cervical cancer biopsies showed a positive reaction. Since some of the genital warts analyzed contained electron microscopically visible viral particles, this was a clearcut suggestion that, most likely, different types of HPV are causative agents of condylomata acuminata. This result prompted a period of analyses of individual virus particle-containing papillomas for agenetic heterogeneity of papillomas; this was established in 1976 [10] and led to the identification of individual types 1 year later [12, 25]. The identification of genital papillomavirus infections turned out to be difficult because of low particle production in genital warts. HPV 6 was identified in 1980 [11] from a rare condyloma with a high particle yield. Two new methodological approaches were of major importance for the subsequent developments: the introduction of gene cloning techniques into the papillomavirus field and the application of hybridization procedures at lowered stringency revealing the relatedness of distinct papillomavirus types, both first used in Peter Howley's laboratory [18]. The use of these methods led to a rapid expansion of identified types of HPVs: today 42 types have been established, and this number will most likely increase further . Gerard Orth in Paris, Stephania Jablonska in Warsaw, and their colleagues identified a large number of HPV types from a rare human condition, epidermodysplasia verruciformis [25]. This syndrome is characterized by an extensive verrucosis and a remarkably high rate of malignant conversions of specific types of papillomas at sunexposed sites. It was of particular interest to demonstrate specific types of HPV, preferentially HPV 5, but also HPV 8 and rarely others, within the carcinomas, although patients with epidermodysplasia verruciformis commonly reveal infections with up to 15 additional types of papillomaviruses. Cancers in this condition therefore appear to represent a particularly interesting example of interactions between a specific virus infection and physical carcinogens such as the ultraviolet part of sunlight. Returning to genital papillomavirus infections, cloning of HPV 6 DNA [6] permitted the identification of a closely related HPV DNA, HPV 11, in genital warts and laryngeal papillomas [13, 14, 22]. Applying conditions of low-stringency hybridization our group identified two additional types of genital papillomavirus infections, HPV 16 and HPV 18, by cloning their DNA directly from cervical cancer biopsies [3, 7]. Subsequently, additional types have been identified, most notably HPV 31 [19] and HPV 33 [2], both somewhat related to HPV 16. The total number of virus types found in the genital tract to date is 14. It appears at present that HPV 6 and 11 are the most prevalent virus types in genital warts, accounting for approximately 90% of all condylomata acuminata and for about one third of all oral papillomas. In contrast, HPV 16 and probably also HPV 18 are found at external genital sites, most commonly in very different lesions, characterized as Bowenoid papulosis or Bowen's disease [16]. The histology reveals marked nuclear atypia and shows characteristics of a carcinoma in situ. All four types of HPV also infect cervical tissue. Typical papillomatous proliferations at the cervix are rare. The most common colposcopically visible lesion is the "flat condyloma" [20]. Although ardently disputed at gynecological and cytological meetings, it is likely that the histology of HPV 6 and 11 lesions differs from that of HPV 16 and 18 [5]. The former viruses induce lesions characterized by a high degree of koilocytosis as a pathognomonic marker for this type of infection. HPV 16 and 18 viruses appear to preferentially induce lesions with marked nuclear atypia, a low degree of koilocytosis, or absence of visible koilocytosis. It is likely that some of the rather confusing data on the histopathology of HPV types result from infections with more than one type of HPV. Probably the majority of histopathologists would not have difficulties in discriminating a Bowenoid lesion from a koilocytotic condyloma at an external genital site. It would be rather surprising if the same type found in both of these lesions induced a uniform histopathological pattern at cervical sites. Kreider and his colleagues [17] recently demonstrated that HPV 11 infections induce changes characteristic of koilocytotic dysplasia upon infection of human cervical tissue heterografted beneath the renal capsule of nude mice. It is anticipated that this technique will establish the causative role of HPVs in cervical dysplasias and therefore point to an etiological role of these agents in a clearly premalignant condition. Since HPV 16 and 18 have been directly isolated from cervical carcinoma biopsies, it is of course important to clarify their role in this type of cancer. HPV 16 DNA is present in approximately 50% of biopsies from cervical, vulvar, and penile cancer. It is also found in some perianal and anal cancers and in a small percentage of oral, tongue, laryngeal, and lung carcinomas. HPV 18 DNA has so far been de tected only in anogenital cancer, occurring in about 20°;., of the biopsies tested. Approximately an additional 10% of these biopsies contain HPV 33,31, or 11 DNA, bringing the total percentage of biopsies with identifiable HPV types to about 80%. It is likely that the majority of additional genital tumors also contain HPV DNA of yet undefined types since bands become visible in blot hybridizations under conditions of lowered stringency. Therefore, it appears justified to summarize this part by stating that the majority of, if not all cervical, vulvar, and penile cancers contain HPV DNA. Similar to cancer biopsies, the majority of cervical cancer-derived cell lines tested so far contain HPV 16 or HPV 18 DNA, among them the well-known HeLa line [3, 34]. The availability of these lines and of primary cervical cancer biopsies permitted an analysis of the state of the viral DNA within the tumor cells. From the results it became evident that cell lines contain exclusively integrated viral DNA; in primary tumors integration is also regularly noted, revealing a monoclonal pattern. Some of the primary tumors contain in addition episomal viral DNA [7]. Precursor lesions, Bowenoid papulosis, and cervical dysplasias appear to contain preferentially nonintegrated viral DNA [8]. The integrational pattern reveals some specificity on the viral side, opening the viral ring molecules most frequently within the E1-E2 open reading frames and thus disrupting the early region [34]. No preferential chromosomal sites have been noted for integration [21]. One line, Caski, contains at least 12 different integration sites in 11 distinct chromosomes. In all cell lines analyzed thus far and in a number of primary tumors, transcription of the persisting HPV DNA has been noted. Commonly, transcripts covering the E6-E7 open reading frames are present [34]. Some of the transcripts are fused to adjacent host cell sequences. It is not clear whether these fusion transcripts play any functional role. Sequencing of cDNA clones of HPV 18 transcripts in three cervical cancer lines revealed the existence of a small intron within the E6 open reading frame [33]. The second E6 ex on is read in a different reading frame, resulting in a putative protein which shows some distant relationship to epidermal growth factor. It is interesting to note that the same splice donor and acceptor sites also exist in HPV 16 and 33 DNA, but are absent in HPV 6 and 1 I DNA. It remains to be seen whether this has any functional significance. The regularity of transcription in HPVpositive cervical cancer cells and the consistent expression of the E6-E7 open reading frames suggest a role of this genetic activity in the maintenance of the transformed state. Integration of viral DNA within E1-E2 with a likely disruption of an intragenomic regulation may represent another event important for malignant conversion. Several recent, still somewhat preliminary studies further emphasize the role of HPV expression in the maintenance of the malignant phenotype. Stanbridge and his co-workers [35] demonstrated that fusion of HeLa cells to normal human fibroblasts or keratinocytes results in a suppression of the malignant phenotype. Loss of chromosomes from the nontransformed donor, apparently in particular that of chromosome no.11 , leads to a reacquisition of malignant growth upon heterotransplantation into nude mice. Since HeLa cells express HPV 18 RNA, it was of obvious interest to analyze HPV 18 expression in the nonmalignant HeLa hybrids as well as in their malignant revertants. The data obtained thus far (E. Schwarz et al., in preparation) indicate that no difference in HPV 18 expression occurs in HeLa cells, in their hybrids with normal cells, or in malignant revertants upon cultivation of these cells in tissue culture. Also under these conditions, no differences are noted in clonability and growth in soft agar. Whereas HeLa cells and malignant revertants continue to express HPV 18 DNA after transplantation into nude mice, initial experiments suggested a complete block of HPV 18 expression in the nonmalignant hybrid lines. Although these data were difficult to interpret due to the invasion of murine cells into the chamber, these and more reccnt studies using differentiation-inducing chemicals in vitro suggest a control of HPV expression at the transcriptional level in nontumorigenic hybrid cells. The interpretation of these data is schematically outlined in Fig. 1. The data point to an intracellular control of HPV expression 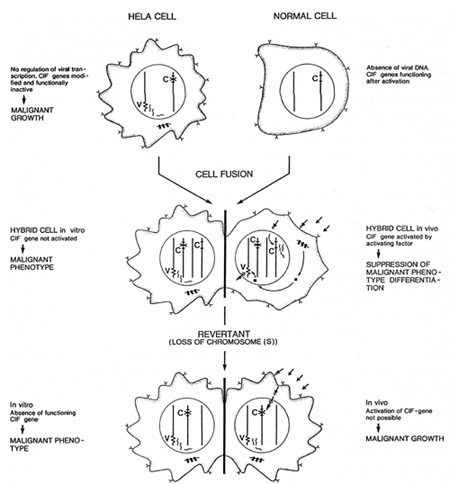
I. Almeida JD, Oricl JD, Stannard LM (1969) Characterization of
the virus found in human genital warts. Microbios 3:225-229 Read more |