|
A. Introduction
The study of 'membrane markers' in human leukaemia has now been
in progress for a decade. Starting from the initial observation
ofL. Borella and colleagues at St. Judes on the sub-types of ALL
[I] a wealth of data has accumulated particularly over the past
few years with the introduction of monoclonal antibodies. Now is
perhaps a good time to appraise the impact of these efforts and
the implications for future research on leukaemia. As Seligmann,
Kersey, myself and others have emphasised on many occasions, the
single most fruitful product of this activity has been the appreciation
of how the cellular heterogeneity of lymphoid leukaemia and lymphoma
mirrors stages of normal differentiation. This clearly arises as
a consequence of three salient features of haemopoietic malignancy:
the restricted or clonal origin [2], the imposition of maturation
arrest, and the broad conservation or fidelity of a qualitatively
normal phenotype [3], The immunological and enzymatic definition
of leukaemic cell phenotypes in relation to their normal counterparts
has direct relevance to clinical problems of differential diagnosis,
patient monitoring and variable prognosis [4]. Immunological features
of ALL subgroups for example are linked to known prognostic features
( e.g. high white cell count in T-ALL) and not surprisingly, therefore,
show a strong correlation with the outcome of chemotherapy [ I,
4-6]. Combinations of markers ( e.g. cell surface antigens and nuclear
terminal transferase [7]) offer the possibility of monitoring leukaemia
and detecting residual, minimal or re-emerging extramedullary disease
(i.e. CNS or testis). The application of a panel of monoclonal antibodies
has been routinely applied in my own laboratory for a national immunodiagnostic
service over a number of years, It is difficult to determine precisely
how useful such a service is; however, I estimate that the phenotypic
data are essential in something like 15% of cases and are useful
or supporting in many more (perhaps the majority). All of this is
clear and undisputed; I would rather emphasise the broader and more
substantial impact which I believe these studies should have. Firstly.
they provide a rational, biological framework for attempts to improve
the efficacy of therapy either by more selective or 'tailored' allocation
of particular regimes to defined leukaemic subgroups or by exploiting
the biological information to design new or more radical strategies,
e.g. monoclonal antibody elimination of leukaemic cells, selective
enzyme inhibition. Secondly, they provide an essential framework
for pursuing the molecular basis of haemopoietic malignancy. Since
cellular oncogenes (or their viral homologues) are probably limited
in number and have some important function in regulating normal
differentiation and/or proliferation, it is of some importance to
search for these genes and the expression and function of their
products in the context of particular leukaemic subtypes and their
normal counterparts; this is indeed already happening (see papers
by F. Wong-Staal and M. A. Lane in this volume). Some of the above
points can be emphasised with reference to the biology of ALL.
B. Heterogeneity and Origins of ALL
Acute lymphoblastic leukaemia can be dissected in a number of subgroups
with exclusive, composite phenotypes, which correlate with prognosis
[4]. More recently, the use of monoclonal antibodies and immunoglobulin
gene probes and the study of maturation induction in vitro has further
elucidated the nature of ALL cells. It is now clear that ALL consists
of two broad
Table 1. Biological features of two ALL
subtypes
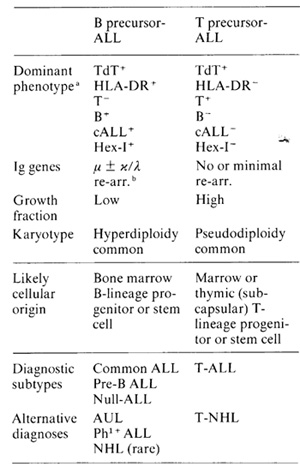
a Serologically defined cell surface antigens or intracellular
enzymes terminal deoxynucleotidyl transferase and hexosaminidase
isoenzyme I (plus charge variants of other acid lysosomal hydrolases.
[42])
b Ig genes (e.g. V. D, J, u heavy chain) re-arranged from germ line
configuration [41]
subtypes, both of which originate in lymphocyte progenitors (Table
I ); one is 'pre-T' or equivalent to thymic precursors of mature
T cells: the other, more common, variant is 'pre-B' or equivalent
to B-cell progenitors and precursors in bone marrow. Within these
two categories subtypes can be defined which broadly reflect sequential
stages of maturation within the 'early' compartments of these two
distinct cell lineages [8-10). Detailed studies on the antigenic
phenotypes of these leukaemias provide no evidence for qualitatively
aberrant gene expression or for a progenitor cell shared by and
exclusive to the T and B lineages. Thus, ALL cells do not express
glycophorin [ II ] or other restricted non-lymphoid markers; neither
do they show concurrent expression on single cells or within a single
leukaemic clone of markers unique to T and B cells. The 'pre- T'
and 'pre-B' categories are also consistent features and although
individual markers may change in relapse [12] there is no shift
between these two subtypes during malignant progression in individual
patients [3]. Normal counterparts of the ALL subtypes with qualitatively
similar phenotypes (excluding karyotype) can be found in bone marrow
[9, 13] and thymus [8. 10], It is of some interest to note that
whereas malignancies of lymphocyte precursors occur predominantly
in children and young patients, malignancies of mature lymphoid
cells (leukaemia, lymphoma, myeloma) are almost exclusively ad u]t
diseases [ ] 3 a]; one interpretation of this correlation and the
similarly striking age associations of other cancers ( e.g. neural
tumours versus epithelial carcinomas) is that they are a reflection
of cell populations (stem cells?) at risk through proliferative
demand at various stages of early development or during prolonged
function (and turnover) in adult life. The simplest interpretation
of this descriptive data is therefore that ALL can originate in
progenitor cells or either the T or B-cell lineage and invariably
sufiers from the imposition of maturation arrest with the conservation
of phenotype 'appropriate' for the particular stage of differentition
in which the leukaemic cells become frozen or stabilised. Whilst
I believe this general conclusion to be manifestly correct there
are some relevant and important qualifications that should be ap
preciated: I. The phenotypes observed are not identical for every
leukaemic blast cell of an jndividual patient. Phenotypic categorisation
reflects the dominant phenotype, but in practice some diversity
can always be detected either with respect to quantity (e.g. antigen
density) or in what appears to be quality. Figure 1 illustrates
one such case, in which one-third of the leukaemic cells have a
different but clearly related phenotype to the other two-thirds.
The interpretation favoured for this intraclonal diversity is that
it reflects in large part the variable stringency of maturation
arrest, i.e. all cells do not appear to be stopped in their tracks
at precisely the same developmental position. Superimposed upon
this maturational control there is also some phenotypic diversity
which is linked to cell proliferation, e.g. expression of the monoclonal
antibody defined receptor for transferrin [ 14 ]. 2. Detailed scrutiny
of ALL phenotypes in relation to their supposed normal counterparts
suggests that they are probably not perfect replicas; an analogy
with the minjmally deviated hepatomas of Potter [15] may be appropriatc.
The "abnormalities" concern some apparent deletions, such as lack
of expression of the E rosette "receptor" or TdT when the remainder
of the composite phenotype dictatcs that they be present or what
can best he described as asynchronies of gene expression, i.e. com
binations of markers which are normally sequentially expressed in
maturation, such as TdT and high-density HLA-ABC in T-ALL [ 10.
16], TdT and .u chains in pre-B ALL[17,40]. 3. Leukaemias with an
identical (non-chromosomal) phenotype to ALL can arise in the pluripotential
stem cell. As reviewed elsewhere [ 18] approximately one-third of
Ph1-positive CGL in blast crisis have the common ALL or B-cell progenitor
phenotype which includes monoclonal antibody defined antigens, selective
enzyme expression and also re-arranged Ig genes (Mulgaard, Gould
and Greaves, unpublished observations). Some adult patients can
present with Ph1 ALL without a clinically evident chronic phase
CGL but may after
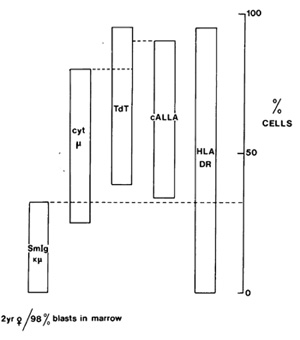
Fig. I. Variable position of "maturation arrest" in cALL.
Bone marrow Iymphoblasts were stained with various combinations
of reagents to analyse phenotypic diversity, e.g. anti-DR, anti-cALL,
anti µ or anti-Ig (chi/lamda ) in combination with TdT; anti-chi
in combination with anti-DR or anticALL
therapy revert to CGL [18]. It is important to note that whereas
B-cell progenitor ALL (e.g. common ALL) is curable with chemotherapy.
blast crises manifest in this cellular compartment are not, although
as expected they may achieve short-term remissions with steroids
[ 19]. This sharp distinction provides an excellent example of the
importance of target cell" biology for understanding clinical outcome
and developing appropriate alternative therapeutic strategies (e.g.
marrow transplants for Phl-positive leukaemia). 4. ALL of either
B or T progenitor type may not be diagnosed haematologically as
ALL. Thus the majority of those rare (~5%) acute leukaemias which
haematologists consider to be acute undifferentiated leukaemia are
usually identifiable as ALL subtypes or more rarely as immature
myeloid cells [4, 20]. Paediatric cases diagnosed as non-Hodgkin
lymphoma may also belong or at least be very closely related to
the two major subtypes of ALL. Conversely, not all cases diagnosed
as ALL may be bona fide ALL. Thus, B-ALL is probably a misnomer:
this relatively mature B-cellleukaemia probably represents a rapidly
disseminating lymphoma [4, 21]. Rare cases of newborn acute leukaemia
diagnosed as ALL may in fact be 'cryptic' erythroleukaemias as assessed
by studies with monoclonal antibodies including antiglycophorin
[11,22]. 5. The maturation arrest imposed in ALL may be reversible.
at least partially in vitro. Thus, some T -ALL cell lines can be
induced by phorbol ester (TPA) to irreversibly modulate their composite
phenotype from that of an immature or thymic variety to that of
a mature T-cell subset [23, 24]. We and others have also been able
to modulate the expression of TdT and cell surface antigen in B-cell
progenitor ALL, although in our experience Ig synthesis cannot be
induced in Ig- ALL despite the presence of re-arranged fA. chain
genes. Our interpretation of this is that in leukaemia and in normal
B-cell differentiation these recombinational genetic events are
inefficient, with most clones failing to achieve a productive or
functional re-arrangement. The observation that maturation arrest
in ALL is reversible as demonstrated previously with other leukaemias
(e.g. Friend virus erythroleukaemia and myeloid leukaemia in rodents.
avian erythroleukaemia and in some human leukaemic cell lines, e.g.
HL-60, K562) carries the important corollarv that maturation arrest,
a central "lesion'; in acute leukaemia, is a regulatory defect which,
although having a genetic, inheritable basis. is reversible in its
phenotypic consequences. C. Is the Conservation of Phenotype Telling
Us Anything Interesting About Leukaemic Cells? It could be argued
that since malignancy involves rare genetic events, it is to be
expected that these will not have catastrophic effects on a cell's
pattern ofgene expression and that the broad fidelity of phenotype
observed in ALL is (a) just what we would expect, and (b) boring
and of no relevance or even downright misleading with respect to
the central issue of what distinguishes a leukaemic cell from normal.
Furthermore, it can always be that the 'critical' gene products
in leukaemia arc not those which we rather arbitrarily elect to
stuliy (so tar) and that a more appropriate screen would reveal
distinct, qualitative and consistent differences between leukaemic
cells and their normal counterparts. These are not unreasonable
views and I am surprised that they are not made more often. I have
favoured a different view initially because it was more interesting
and subsequently because I believe it is supported by data. That
is that the expression of qualitatively normal phenotype or pattern
of gene expression is an integral and essential feature of most
if not allleukaemias and other malignancies. Qualitative abnormalities
(e.g. new or lost antigens, altered glycolipids, altered drug recognition)
may occur and indeed have some selective advantage with malignant
progression and treatment: however, they need not be considered
as essential components of the malignant state. In the context of
ALL, therefore, and as suggested some years ago [25, 26] a qualitatively
normal lymphoid progenitor cell phenotype which is normally only
transiently expressed on proliferating cells quite compatible with
leukaemic
Table 2. Structure, genetics and function
of ALL-associated membrane proteins identified by monoclonal antibodies
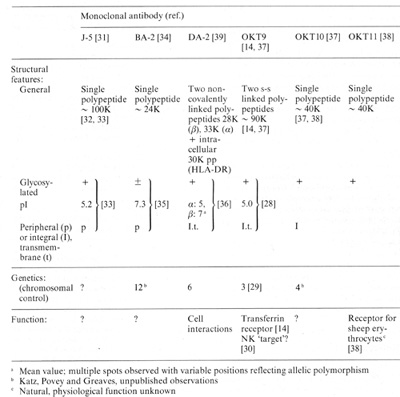
a Mean value: multiple spots observed with variable positions
reflecting allelic polymorphism
b Katz, Povey and Greaves, unpublished observations
c Natural, physiological function unknown
cell behaviour and only requires that the genetic change provoking
clonal selection effectively, uncouples proliferation from maturation.
This view accords with recent molecular studies which reveal the
central role of normal genes (c-onc) or their inserted viral (v-onc)
homologues which may facilitate clonal advantage via amplification
or excessive promotion ([27] and various papers in this volume).
There is no evidence to date that qualitatively altered gene products
are involved (An important example of such an alteration has however
recently been reported [43] ) Much emphasis therefore rests on quantitative
aspects of c-onc expression. Even this phenotypic distinction between
leukaemic and normal cells could be small or perhaps only evident
in the time frame, i.e. equivalent normal cells may express similar
levels of c-onc gene products but only transiently.
D. Epilogue
Several of the ALL-associated membrane antigens have now been biochemically
characterised and their control mapped to particular chromosomes
(Table 2). Whether any of these proteins has any important regulatory
role in differentiation or are even c-onc gene products is at present
unknown. One of these structures does have a definite function.
The monclonal antibody OKT9 identifies the transferrin receptor
[14]; this observation has enabled rapid progress to be made in
the biochemica! studies of this receptor [28] and also facilitated
the mapping of controlling (presumably structural) genes to chromosome
3 [29]. We have also suggested that the transferrin receptor may
serve as a common 'target' structure on malignant and normal cells
for so-called natural killer (NK) cells [30]. There are still many
gaps in our understanding of lymphoid malignancy and of normal lymphopoiesis.
Compared with myelopoiesis for example (see paper by Metcalf in
this volume) we have little insight into soluble regulators of early
lymphocyte development. Despite these limitations lymphoid malignancy
in humans provides, I believe, an excellent example of a disease
whose molecular, cellular and clinical complexity can be best understood
in relationship to normal cellular differentiation.
References
I Sen L, Borella L ( 1975) Clinical im portance of Iymphoblasts
with T markcrs in childhood acute leukemia. N Engl J Med 292: 828-832
2. Fialkow PJ, Denman AM, Singer J, Jacobson RJ, Lowenthal MN (1978)
Human myeloproliferative disorders. clonal origin in pluripotential
stem cells. In. Clarkson B, Marks PA, Till JE (eds) Differentiation
of normal and neoplastic hemopoietic cells. Cold Spring Harbor,
New York, pp 131-144
3. Greaves MF (1982) 'Target' cells, cellular phenotypes and lineage
fidelity in human leukaemia. J Cell Physiol Suppl I: 113-126
4. Greaves MF (1981) Analysis of the clinical and biological significance
of lymphoid phenotypes in acute leukemia. Cancer Res 41:4752-4766
5. Dow L W, Borella L, Scn L, Aur RJA, George SL, Mauer AM, Simone
JV (1977) Initial prognostic factors and Iymphoblasterythrocyte
rosette formation in 109 children with acute lymphoblastic leukemia.
Blood 50.671-682
6. Greaves MF, JanO.5sy G, Pcto J, Kay H ( 1981) Immunologically
dcfined subclasscs of acutc lymphoblastic leukaemia in children.
their relation5hip to prcsentation features and progno5is. Br J
Haematol 48179-197
7. Janossy G, Bollum FJ, Bradstock KF, AshIcy J (1980) Cellular
phcnotypes of normal and leukemic hemopoietic cclls determined by
analysis with 5elected antibody combi nations. Blood 56. 430-441
8. Reinherz EL, Kung PC, Goldstein G, Levey RH, Schlossman SF (
1980) Discrete stages or human intrathymic differentiation. analysis
or normal thymocytes and leukemic Iymphoblasts of T lineage. Proc
Natl Acad Sci USA 771588-1592
9. Greaves MF, Delia D, Robin,5on J, Sutherland R, Newman R (1981)
Exploitation or monoclonal antibodies: A 'Who's who' or haemopoietic
malignancy. Blood Cells 7:257-280
10. Greaves, MF, Rao J, Hariri G, Verbi W, Catovsky D, Kung p, Goldstein
G (1981) Phenotypic heterogeneity and cellular origins of T -cell
malignancies. Leukcmia Res 5:281-299
II. Greaves MF (1981) Monoclonal antibodies as probcs ror leukaemic
heterogeneity and haemopoietic differentiation. In: Knapp W. (ed.)
Leukemia markers. Academic, New York, pp 19-32
12. Greaves MF, Paxton A, Janossy G, Pain C, Johnson S, Lister TA
(1980) Acute lymphoblastic leukaemia associated antigen III. Alterations
in expression during treatment and in relapse. Leukemia Res 4.1-14
13. Greaves MF, Robinson JB, Delia D, Ritz J, Schlossman S, SieffC,
Goldstein G, Kung P, Bollum r, Edwards P ( 1981) Comparative antigenic
phenotypes or normal and leukemic hcmopoietic precursor cells analysed
with a 'library' of monoclonal antibodies In. Neth R, Gallo RC,
Graf T, Mannwcilcr K, Winklcr K (eds) Modern trends in human leukemia
4. Springer, Berlin Heidelberg New York, pp 296-304 (Haematology
and blood transfusion, vol 26)
13 a. Greaves MF (to be publishcd) Subtypes of acute lymphoblastic
leukaemia: implications for the pathogenesis and epidemiology of
leukaemia. In: Magrath I, Ramot B (eds) The influence of the environment
on leukaemia and lymphoma subtypes. Natl Cancer Inst Monogr
14. Sutherland R, Delia D, Schneider C, Newman R, Kemshead J, Greaves
MF (1981) Ubiquitous, cell surface glycoprotein on tumour cells
is proliferation-associated receptor for transferrin. Proc Natl
Acad Sci USA 78:4515-4519
15. Potter VR ( 1978) Phenotypic diversity in experimental hepatomas:
the concept of partially blocked ontogeny. Br J Cancer 38.1-23
16. Bradstock Kf', Janossy G, Bollum FJ, Milstein C (1980) Anomalous
gene expression in human thymic acute lymphoblastic leukaemia (Thy-ALL).
Nature 284.455-457
17. Greaves MF, Verbi W, Vogler L, Cooper M, Ellis R, Ganeshaguru
K, Hoffbrand V, Janossy G, Bo!lum FJ (1979) Antigenic and enzymatic
phenotypes of the pre-B subclass of acute lymphoblastic leukaemia.
Leukemia Res 3.353-362
18. Greaves MF ( 1982) 'Target' cells, differentiation and clonal
evolution in chronic granulocytic leukaemia: A 'model' for understanding
the biology of malignancy. In: Shaw MT (ed) Chronic granulocytic
leukaemia. Praeger, New York, pp 15-47
19. Greaves MF (1981) Biology of acute lymphoblastic leukaemia.
16th Annual Guest Lecture: Leukaemia Research Fund Publ
20. Greaves MF, Bell R, Amess J, Lister T A (to be published) What
is 'undifferentiated' leukaemia?
21. Magrath IT, Ziegler JL (1980) Bone marrow involvement in Burkitt-s
lymphoma and its relationship to acute B-cell leukemia. Leukemia
Res 4.33-60
22. Greaves MF, Sieff C, Edwards P (1983) Monoclonal anti-glycophorin
as a probe for erythroleukaemia5. Blood (in press)
23. Nagasawa K, Mak TW(1980) Phorbol esters induce differentiation
in human malignant T Iymphoblasts. Proc Natl Acad Sci USA 77.2964-2968
24. Delia D, Greaves M, Newman R, Sutherland R, Minowada J, Kung
P, Goldstein G (1982) Modulation of T leukaemic cell phenotype with
phorbol ester. lnt J Cancer 29.23-31
25. Greaves MF, Janossy G (1978) Patterns of gene expression and
the cellular origins of human leukaemia. Biochim Biophys Acta 516:193-230
26. Greaves MF (1979) Tumour markers, phenotypes and maturation
arrest in malignancy. A cell selection hypothesis. In Boelsma E,
Riimke P (eds) Tumour markers. Elsevier, Amsterdam, pp 201-211
27. Varmus H (1982) Recent evidence for oncogenesis by insertion
mutagenesis and gene activation. In. Greaves M (ed) Leukaemia cell
differentiation. Cancer Surveys, vol 2. ICRF, London, pp 309-320
28. Schneider C, Sutherland R, Newman R, Greaves M (1982) Structural
features of the cell surface receptor for transferrin that is recognised
by the monoclonal antibody OKT9.J Bioi Chem 251:8516-8522
29. Goodfellow PN, Banting G, Sutherland R, Greaves M, Solomon E,
Povey S (1982) Expression of the human transferrin receptor is controlled
bye gene on chromosome 3. assignment using the species specificity
of a monoclonal antibody. Somatic Cell Genet 8.197-206
30. Vodinclich L, Sutherland DR, Schneider C, Newman R, Greaves
MF (1983) The receptor for tran5ferrin may be a 'target' structure
for natural killer cells. Proc Natl Acad Sci (in press)
31. Ritz J, Pesando JM, Notis-McConarty J, Lazarus H, Schlossman
SF ( 1980) A mono clonal antibody to human acute lym phoblastic
leukaemia antigen. Nature 283.583-585 32. Sutherland R, Smart J,
Niaudet P, Greaves MF (1978) Acute lymphoblastic associated antigen.
11. Isolation and partial characterization. Leukemia Res 2: 115-126
33. Newman RA, Sutherland R, Greaves Mf' ( 1981) The biochemical
characterization of a cell surface antigen associated with acute
lymphoblastic leukemia and lymphocyte precursors. J ImmunoI126:2024-2030
34. Kersey JH, LeBien TW, Abramson CS, Newman R, Sutherland R, Greaves
M (1981) p24. a human leukemia-assocjated and Iymphohemopoietic
progenitor cell surf'ace structure identified with monoclonal antibody
J Exp Med 153.726- 731
35. Newman RA, Sutherland DR, LeBien TW, Kersey JH, Greaves Mf'
(1982) Biochemical characteriZiltion of a leukaemia-associated antigen
(p24) defined by the monoclonal antibody BA-2. Biochim Biophys Acta
701:318-327
36. Newman R, Greaves MF (1982) Characterisation of HLA-DR on leukaemic
cells. Clin Exp ImmunoI50.41-50
37. Terhorst C, Van Agthovan A, LeCJair K, Snow P, Reinherz E, Schlossman
S ( 1981 ) Biochemical studies of the human thymocyte cell-surf'ace
anti~ens T6, T9 and TIO. Cell 23.771-780 v
38. Verbi W, Greaves MF, Schneider C, Koubek K, Jano55y G, Stein
H, Kung P, Goldstein G (1982) Monoclonal antibodies OKTIO and OKTIIA
have pan T reactivity and b1ock sheep erythrocyte 'receptors'. Eur
J Immuno11281-86
39. Brodsky FM, Parham P, Barnstable CJ, Crumpton M, Bodmer WF (1979)
Hybrid myeloma monoclonal antibodies against MHC products. Immunol
Rev 47.3-61
40. Vogler LB, Crist WM, Bockman DE, Pearl ER, Lawton AR, Cooper
MD (1978) Pre-B cellleukemia. a new phenotype of childhood lymphoblastic
leukemia. N Engl J Med 298.872-878
41. Korsmeyer SJ. Hieter PA. Ravetch. JV. Poplack DG. Waldmann TA.
Leder P (1981) Developmental hierarchy of immunoglobulin gene rearrangements
in human leukemic pre-B cells. Proc Natl Acad Sci USA 78.7096-7100
42. Dewji N. Rap5on N. Greaves M, Ellis R (1981) Isoenzyme profiles
of lysosomal hy drolases in Ieukaemic cells. Leukemia Res 5.19-27
43. Reddy EP, Reynolds RK, Santos E, Barbacjd M (1982) A point mutation
is responsible for the acquisition of transforming properties by
the T24 human bladder carcinoma oncogene. Nature 300.149-152
|