|
The University of Texas M.D. Anderson Cancer Center,
Dept. of Pediatrics, Houston, Texas 77030, USA
Forty years ago, Farber and associates described temporary remissions
of acute leukemia in children produced by folic acid antagonists
[13]. This ignited the hope that this most frequent and always fatal
childhood cancer might be curable by drugs. Twenty years ago, Aur
and associates completed accession of patients to total therapy
study V, the first treatment protocol to result in 50% cure of acute
lymphoid leukemia (ALL) [3]. Their results stand 20 years later
(Fig. 1 ), and have been reproduced throughout the world in many
thousands of children [6]. More important, recent national vital
statistics of the United States and the United Kingdom indicate
a 50 % reduction in childhood leukemia mortality [4, 29]. Further,
the cured children generally enjoya normal life-style without need
for medication. In the past 20 years, efforts have been directed
at improving the cure rate of ALL while simplifying curative treatment,
reducing its side effects, and improving its availability and accessibility.
In a Stohlman Lecture at Wilsede 10 years ago the following statement
was made [32]: -The most significant opportunity for improving the
treatment of acute lymphoid leukemia in the past five years has
been its biological and clinical classification by immunological
cell surface markers. This allows species identification of the
leukemia cells, the first step toward developing specific cytocidal
or cytostatic therapy. The purpose of this communication is to review
progress in immunophenotypespecific therapy of ALL, to discuss some
alternate methods of guiding treatment, and to introduce the notion
of genotypespecific chemotherapy of ALL.
A. Immunophenotype-Specific Therapy of ALL
I. Historical Perspective
When the first effective drugs were used to treat acute leukemia
it became apparent that some cases were more responsive than others
[12]. Methotrexate, prednisone, or mercaptopurine were most likely
to produce remissions in children with ALL. Adults with ALL were
less likely to experience remission. Both children and adults with
acute nonlymphoid
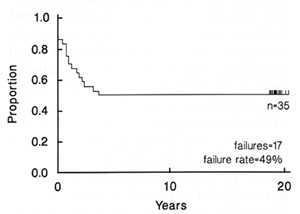
Fig. 1. Event-free survival (EFS) of 35 consecutive children
with acute lymphoid leukemia admitted to St. Jude Children's Research
Hospital from December 1967 to June 1968. Approximately one-half
remain continuously free of leukemia for 20 years and off therapy
for 18 years. Update of [3], kindly provided by Gaston Rivera
leukemia (ANLL) had few remissions with these agents.
Some hematologists concluded that chemotherapy was of little use
in adult acute leukemia and was perhaps better withheld in ANLL,
in children as well as adults. With the introduction of daunorubicin
and cytarabine in the 1960s it became apparent that these drugs
were highly active in the majority of patients with ANLL, especially
when combined [18]. On the other hand, their value in childhood
ALL was not so apparent. The concept of species-specific therapy
was thus evolved and it became customary to utilize prednisone,
vincristine, methotrexate, and mercaptopurine as the primary drugs
for ALL, and daunorubicin and cytarabine as the mainstay of treatment
of ANLL.
II. Species-Specifie Therapy of T -Cell ALL
When T-cell ALL was first defined it was noted that children with
this disease had short remissions and high mortality compared with
children who had non- TALL [43]. These observations were generally
confirmed by others. However, in mice it was demonstrated that cyclophosphamide
and cytarabine were more effective in AKR leukemia, a T -cellline,
and Sullivan et al. suggested that cytarabine was specifically effective
in human T -cell lymphoma/leukemia [42, 47]. A comparative study
in children with ALL in remission demonstrated that the cure rate
of T -cell ALL approached that of non- T ALL when the T -cell patients
received cyclophosphamide and cytarabine in addition to methotrexate
and mercaptopurine [26]. On the other hand, the cyclophosphamide
and cytarabine provided no curative benefit, only additional toxicity,
to children with non- T ALL receiving methotrexate and mercaptopurine.
Thus, it became clear that immunophenotype of ALL was important
in selecting and scheduling curative drug therapy. The importance
of immunophenotype-specific chemotherapy of T -cell lymphoma/leukemia
was confirmed in a recent Pediatric Oncology Group study [1]. With
a treatment plan that emphasizes the use of cytarabine, cyclophosphamide,
Adriamycin, and teniposide, and excludes systemic methotrexate,
actuarial event-free survival for 94 children with T -cell ALL is
71°1. at 18 months. Since most relapses of T-cell ALL occur within
18 months this is a meaningful figure.
III. Species-Specific Therapy of B-Cell ALL
When B-cell ALL was defined its rapidly fatal course despite chemotherapy
was noted and confirmed [15]. However, two reports indicate that
distinctive treatment plans emphasizing the use of cYclophosphamide,
the most active agent in childhood B-celllymphoma/ ALL, and a concentrated,
relatively brief multipledrug program, result in a 40% cure rate
[14, 30]. A Pediatric Oncology Group study appears to be confirming
these observations (Bowman, personal communication).
IV. Species-Specific Therapy of Non- T Non-B ALL
The question rises whether speciesspecific therapy of subclasses
of non- T non-B ALL might be appropriate. As with T -cell ALL and
B-cell ALL, the first suggestion of the need for specific therapy
is the appearance of an association between immunophenotype and
prognosis on a given treatment regimen. Just as T -cell ALL and
B-cell ALL demonstrated short remissions and very high mortality
in early treatment programs, two immunophenotypic species of non-
T non-B ALL have had less favorable courses in more recent studies.
First is the "null" or pre-B lymphoid/monocytoid species associated
with age less than 1 year, low CALLA antigen, chromosomal translocations
involving chromosomc 11, band
Table I. Species-specific therapy, non- T, non-B ALL,
treatment plan

The systemically administered mercaptopurine, methotrexate, cytarabine,
cyclophosphamide, and etoposide are given in maximum tolerated dosage,
using clinical status, absolute phagocyte count, and mean corpuscular
volume as guides
q 23, presence of myeloid antigens, and monocytoid characteristics
by electron microscopy and cell culture [23], Second is pre-B ALL,
which demonstrates cytoplasmic immunoglobulin and is sometimes associated
with a t(1;19) chromosomal translocation [35], A species ofT-cell
ALL that demonstrates CALLA antigen is reported to have a cure rate
between that of T-cell ALL and common ALL on traditional therapy
[9]. At UT MD Anderson Cancer Center a pilot protocol was designed
and initiated for children newly diagnosed with non- T non-B ALL
that provides different periodic consolidation therapy for four
different species: common (early pre-B CALLA+), null (early pre-B
lymphoid/ monocytoid), early pre-B CALLA+ and thymic antigen + ,
and pre-B (Table 1). Each of the four regimens utilizes periodic
consolidation drugs and drug schedules that are currently believed
to be most effective for these specific subclasses, while retaining
a core of conventional continuation therapy with daily mercaptopurine,
weekly methotrexate, pulses of prednisone, vincristine and asparaginase,
and periodic triple-intrathecal therapy. Early results suggest the
feasibility of this pilot protocol. Of 26 consecutive children registered
in the past 18 months, 24 developed complete remission. None have
experienced relapse yet. In summary, immunophenotypespecific selection
and scheduling of chemotherapy has proven to be important for increasing
the cure rate of T -cell and B-cell ALL. It may also be applicable
to upgrading the curability of null ALL and pre-B ALL as well. Almost
as important, immunophenotype-specific therapy allows one to exclude
nonessential antineoplastic drugs from the combination chemotherapy
regimens of ALL, thus avoiding unnecessary immediate and long-term
toxic hazards. The prime example is hyperdiploid common ALL, which
is highly curable with methotrexate and mercaptopurine continuation
chemotherapy [6, 49]. There is no evidence that addition of anthracyclines
or alkylating agents improves its cure rate [5]. Therefore, there
is no reason to expose these highly vulnerable pre-school children
to the risks of anthracyeline cardiomyopathy or cyclophosphamide-induced
bladder carcinoma [27, 31].
B. Selection and Scheduling Chemotherapy by "Prognostic Factors"
It was recognized decades ago that initial white blood cell count
was predictive of response to leukemia chemotherapy [51]. Subsequently,
other factors were identified and the term "high risk for treatment
failure" was coined for patients with ALL who had such features
[2]. It was suggested that more extensive remission induction chemotherapy
be administered to such patients. Since then, terms such as "standard
risk," "low risk," and "high risk" have become popular to define
prognostic categories of patients with ALL and to select and schedule
their chemotherapy [46]. In general, patients with "high-risk" ALL
are given more drugs in higher dosage, particularly such agents
as anthracyclines, alkylating compounds, and epipodophyllotoxins.
Patients with "low-risk" ALL are given fewer drugs in lesser dosage,
primarily corticosteroid, vinca alkaloid, and antimetabolites. In
some treatment programs the decision to use cranial irradiation
is based on "risk group" [46]. The problem with using prognostic
factors to select therapy is that they are artifacts of data analysis
and treatment [33, 34]. More aggressive and rapidly proliferating
ALL tends to relapse early; less aggressive and slowly proliferating
ALL tends to relapse late. When complete remission duration is used
as the criterion for assessing prognostic factors undue weight is
given to features associated with remission duration rather than
to the true measure of efficacy of therapy, cure, as represented
by the plateau of continuous complete remission. This problem with
the use of prognostic factors could be corrected by using cure rate
instead of remission duration to calculate prognostic variants.
However, the more important issue is treatment artifact. All leukemias
are fatal when untreated. Survival and cure depend on the administration
of appropriate drugs in appropriate schedules. For example, when
T -cell ALL was treated with conventional non- TALL chemotherapy
it had a rapidly fatal course in most patients [26]. Features associated
with T -cell ALL such as thymic mass, male sex, high white cell
count, and older age were calculated to be "highrisk" or "bad-prognosis"
factors. With appropriate chemotherapy ofT -cell ALL these "risk
factors" largely disappear. In conclusion, there is no evidence
that one type of ALL is inherently more lethal than another. All
are equally lethal. Cure of ALL is solely a matter of developing
and selecting the appropriate drug regimens for each specific type
of ALL. The use of prognostic factors to guide leukemia therapy
should be abandoned because it is based on artifacts and can give
rise to erroneous conclusions.
C. All-lnclusive Multiple-Drug Chemotherapy for All ALL
Another method of selecting therapy for ALL is to avoid selection,
but to give all patients all active antineoplastic drugs without
regard to immunophenotypic species [37]. This approach carries multiple
problems. Unlike antibiotics, most antineoplastic drugs have overlapping
short-term side effects. Administration of one drug usually interferes
with the dosage of the other. If minimally effective or noneffective
drugs are included in a combination, the dosage of the more effective
drugs generally must be reduced. If numerous drugs with overlapping
toxicities are utilized it is possible that the most effective drug
or drugs may be given at minimally effective dosages and their benefit
compromised or lost. Exposure to suboptimal dosage of drugs is an
important mechanism of developing resistant cell lines in vitro
and could be a mechanism in vivo. In some all-inclusive multiple-drug
regimens, drugs or drug combinations are alternated in order to
minimize reduction of drug dosages [37]. The problem with this technique
is that the leukemia, in effect, may be untreated or minimally treated
during those intervals when drugs of minimal or no efficacy for
that particular leukemia are being given. One might postulate the
possibility of resurgence of leukemia cell proliferation during
such periods of minimally effective or noneffective therapy. A theoretical
objection to the use of multiple drugs is the possibility of antagonistic
interactions that might subtract from the efficacy of a given drug
[21]. Little is known about subtractive drug interactions in human
cancer chemotherapy. One would assume that the risk of such interactions
would increase geometrically with linear increase in the number
of drugs administered. A major concern of cancer chemotherapy in
children is the prospect of serious long-term sequelae. As noted
previously, of special concern are the anthracyclines and the alkylating
agents. In one study of children surviving ALL, 55% of those who
had received doxorubicin demonstrated abnormal left ventricular
function and/or afterload by echocardiography [27]. Cyclophosphamide
not only produces sterility but carries a 10% risk of urinary bladder
carcinoma 12 years later [31]. To exemplify this concern, it is
known that children with hyperdiploid common ALL have a 70% or greater
cure rate without alkylating agents or anthracyclines [6, 49]. The
only comparative studies reported have failed to demonstrate that
these agents contribute to the cure of common ALL in first remission
[5]. For these reasons they should be avoided in children with hyperdiploid
common ALL who are newly diagnosed or in first remission. The same
can be said for any drug with demonstrated serious sequelae that
has failed comparative testing for its value in contributing to
the cure of a specific type of ALL. A final objection to the all-inclusive
multiple-drug chemotherapy approach is its excessive complexity
and eost. This tends to limit the availability and accessibility
of curative leukemia therapy to more privileged patients and more
privileged nations. The objective of leukemia therapy is to reduce
national and world leukemia mortality, not only that of well financed
medical centers.
D. Genotype-Specific Therapy of ALL
I. Acute Leukemias Are Genetic Disorders of Hematopoietic Cells
The most important advance in leukemia therapy in the past 10 years
is the renewed realization that leukemias are genetic disorders
of hematopoiesis [34, 38, 41]. Their abnormal morphology, immunophenotype,
growth, and function are all reflections of their genetic abnormalities.
This opens a pathway of drug therapy specific to their genetic properties,
aimed at converting their genetic advantages to liabilities. The
evidence that acute leukemias are genetic disorders is convincing
[34]. The risk of leukemia is increased in certain constitutional
genetic disorders such as Down's, Fanconi's, and Bloom's syndromes
and in persons exposed to mutagens such as ionizing irradiation.
The morphology of leukemia cells tends to be disorderly and asynchronous,
reflecting disordered genetic expression. Chromosome morphology
is disturbed in most acute leukemias [41]. Nonrandom chromosome
abnormalities are associated with specific types of acute leukemia,
such as the t( 1; 19) translocation in pre- B ALL, the t(8;14) in
B-cell ALL, and the t(15;17) in acute promyeloid leukemia [7, 35,38].
Immunophenotypic and molecular genetic disorders are also prevalent
in acute leukemias [20, 34, 45]. Some ALLs express surface antigens
characteristic of B-cell and T -cell lineage simultaneously. Early
pre-B-(common) ALL often demonstrates rearrangement of genes encoding
the T -cell receptor while T -cell ALL may show gene rearrangement
for immunoglobulins. It is now obvious that ALLs do not have true
B-Iymphocyte or T -lymphocyte lineage. Their genetic and phenotypic
immunological markers are merely further reflections of their underlying
genetic disorders. ALL is a genetic, not an immunological, disease.
The most recent evidence that acute leukemias are genetic disorders
is the discovery of overexpression of certain oncogenes in some
cases, for example, c-myc in B-cell ALL and c-sis in acute megakaryocytic
leukemia [7, 48].
II. Chemotherapy May Cure Acute Leukemia by Genetic Mechanisms
Although chemotherapy appears to induce remissions of acute leukemIa
by direct cytolytic effects, it is possible to speculate that cures
result from genetic alteration during chemotherapy [34]. Curative
drugs such as methotrexate, cytarabine, cyclophosphamide, daunorubicin,
and etoposide alter DNA structure as well as synthesis, while drugs
without direct effect on DNA such as prednisone, vincristine, and
asparaginase do not appear to be curative. . Secondly, curative
chemotherapy elIminates genetically disturbed hematopoiesis but
spares the capacity for genetlcally normal hematopoiesis [34]..
The best example is the lymphoblastIc and lymphocytic hyperplasia
noted in the bone marrow of children with ALL after cessation of
chemotherapy. Sometimes the frequency of CALLA + Iymphoblasts in
these children is sufficient to cause confusion with relapse. Finally,
the curative capacity of chemotherapy is strongly related to the
genotype of the Icukemia [34, 41]. For examplc, methotrexate and
mercaptopurine is a highly curative drug combination in hyperdiploid
common ALL, but not in common ALL with a t(9;22) translocation [45,49].
Daunorubicin and cytarabine is more often curative in acute myeloid
leukemia (AML) with a t(8;21) translocation than in AML wIthout
this translocation [41]. It is possible that leukemia chemotherapy,
when it is curative, is more specific in affecting the genetic mechanism
or genetic survival of leukemia strains than we have recognized.
III. Rationale for Genotype-Specific Therapy of ALL
The basis for attempting to target chemotherapy of ALL to its genotypic
characteristics is severalfold. First is the convincing evidence
that acute leukemias are genetic disorders of hema.topoietic cells
[34]. Their morphology, Immunological markers, growth rate, and
other phenotypic properties are reflectIons of their specific genetic
disorders. Secondly, genetic properties arc the most significant
variables in curability by a given therapeutic regimen. [6, 49]..
This indicates that therapeutIc regImens should be varied in accordance
with the genetic properties of the leukemias in order to achieve
optimal cure rates. For example, common ALL with a t(9;22) translocation
needs to be treated differently than common ALL with hyperdiploidy
in order that the t(9;22) variety becomes as curable as the hyperdiploid
type. Thirdly, the current practices of altering chemotherapy regimens
in accordance with morphology (ALL vs. ANLL), immunophenotype (T
cell vs. B cell), and aggressiveness (white blood cel1 count) in
fact do recognize genotypic properties because all these features
reflect the genetic disorders. It would appear more rational to
aim treatment directly at the genetic disorders that underIy these
features as we learn to define these disorders more precisely. Finally,
as noted above, there is reason to speculate that chemotherapy .produces
remissions by direct cytotoxicity but cures by genetic alteration.
IV. Relationships Between Genotype and Drug Efficacy in ALL
The relationships between the known genotypes of acute lymphoid
leukemias and what appear to be the most effective drugs and drug
combinations for curing them are summarized in Table 2. The data
are yet fragmentary, only the beginning of an approach at targeting
drug therapy to the genetic disorders of the
Table 2. Genotype and drug curability, acute lymphoid
leukemia

Many of the molecular genetic and drug data are unconfirmed or
speculative
leukemias rather than to the phenotypic features that reflect the
genetic disorders. As breakpoints of chromosomal translocations
are defined in molecular terms and it becomes possible to classify
leukemias as specific molecular genetic disorders it is to be expected
that leukemias without apparent chromosomal rearrangements will
be shown to have rearrangements of genes similar to those that do
have the chromosomal changes. This has already been described in
adult-type chronic myeloid leukemia where cases without the typical
t(9;22) translocation have the same bcr-abl rearrangement that occurs
in those with the translocation [24, 44]. As the acute leukemias
become better defined in molecular genetic terms it seems plausible
that genotype-specific therapy will become more apparent and feasible.
E. Summary
In the past 10 years immunophenotyping of ALL has been demonstrated
to be useful for selecting and scheduling chemotherapy. Different
drug regimens are now used for T -cell and E-cell ALL than for non-
T non-E ALL with the result that survival and cure of T -cell and
E-cell ALL have been considerably improved. The use of different
drug regimens for different immunophenotypic varieties of non- T
non-E ALL is being tested. "Prognostic factors" of ALL are artifacts
of data analysis and treatment and should no longer be used for
guiding treatment. The administration of all-inclusive multiple-drug
therapy to all patients with ALL regardless of species should also
be abandoned. Minimally effective drugs can interfere with dosage
and continuity of more effective drugs, and can result in side effects
and sequelae that increase the mortality and morbidity of treatment.
Since acute leukemias are genetic disorders of hematopoiesis the
future direction of leukemic therapy is toward genetic targeting.
References
1. Amylon M, Murphy S, Pullen Jet al. (1988) Treatment of lymphoid
malignancics according to immune phenotype. Preliminary results
in T -cell disease (Abstr). Proc Am Soc Clin Oncol 7.225
2. Aur RJA, Simone JV, Pratt CB et al. (1971) Successful remission
induction in children with acute lymphocytic leukemia at high risk
for treatment failure. Cancer 27.1332-1336
3. Aur RJA, Simone JV, Hustu HO et al. (1971) Central nervous system
therapy and combination chemotherapy of childhood lymphocytic leukemia.
Blood 37.272-281
4. Birch JM, Marsden HB, Jones PH et al. (1988) Improvements in
survival from childhood cancer. results of a population based survey
over 30 years. Br Med J 296.1372 1376
5. Camitta BM, Pinkcl D, Thatcher Get al. (1980) Failure of early
intcnsive chcmotherapy to improve prognosis in childhood acute lymphocytic
leukemia. Med Pediatr Oncol 8: 383-389
6. Crist WM, Furman W, Strother D et al. (1987) Acute lymphocytic
Ieukemia in childhood Immunologic marker, cylogcnetic, and molecular
studies. South Med J 80: 841-847
7. Croce CM (1986) Chromosomc translocations and human cancer. Cancer
Res 46.6019-6023
8. Denny CT, Hollis GF, Hecht F et al. (1986) Common mechanism of
chromosome inversion in R- and T -cell tumors. Relevance to lymphoid
development. Science 234. 197- 200
9. Dowell BL, Borowitz MJ, Boyett JM et al. (1987) Immunologic and
clinicopathologic features of common acute lymphoblastic leukemia
antigen-positive childhood T cellleukemia. Cancer 59.2020-2026
10. Dube ID, Raimondi SC, Pi D et al. (1986) A new translocation,
t(10;14) (q24;qI1), in T cell neoplasia. Blood 67.1181-1184
11. Erikson J, Finger L, Sun L ct al. (1986) Deregulation of c-myc
by translocation of the alfa-Iocus of thc T -cell rcceptor in T
-cell leukemias. Science 232.884-886
12. Farber S, Toch R, Sears EM, Pinkel D ( 1956) Advances in chemotherapy
of cancer in man. Adv Cancer Res 4: 1- 71
13. Farber S, Diamond LK, Mercer RD ct al. (1948) Tcmporary rcmissions
in acute letikcmia in childrcn produced by folic acid antagonist,
4-aminoptcroyl-glulamic acid (aminopterin). N Engl J Med 238.787
-793
14. Feickert HJ, Göbel U, Ludwig Wet al. (1987) Childhood acute
lymphoblastic Ieukemia of B-cell typc: Trials ALL-BFM 81 and ALL-BFM
83 (Abstr). Proc Am Soc Clin Oncol 6.149
15. Flandrin G, Brotict JC, Daniel MT et al. (1975) Acute leukemia
with Burkitt's tumor cells. A study of six cases with special reference
to Iymphocylc stirface markers. Blood 45: 183-188
16 Finger LR, Harvey RC, Moore RC ct al. ( 1986) A common mechanism
of chromosomal translocation in T- and B-cell neoplasia. Science
234: 982-985
17. Frankel LS, Ochs 1, Shuster Jet al. (1987) Pilot protocol improves
remissions for infant leukemia and provides detailed laboratory
characterization (Abstr). Proc Am Soc Clin Oncol 6.161
18. Gale RP (1979) Advanccs in the treatment of acute myelogcnous
leukcmia. N Engl J Med 300:1189-1199
19. Goyns MH, Hann IM, Stewart Jet al. (1987) The c-els-1 proto-oncogene
is rearranged in some cases of actite lymphoblastic leukaemia. Br
J Cancer 56.611-613
20. Hurwitz CA, Loken MR, Graham ML, et al. (1988) Asynchronous
antigen expression in B lineage acute lymphoblastic leukemia. Blood
72: 299-307
21. Jolivet J Cole D, Holcenberg JS et al. (1984) L-asparaginase
(L-ASP) antagonism of methotrexate (MTX) cytotoxicity. An alternative
explanation (Abstr). Pro ceedings of thc American Association for
Cancer Research 25.309
22 Kancko Y, Mascki N, Takasaki Net al. ( 1986) Clinical and hematologic
characteristics in acutc leukemia with I1q23 translocations Blood
67.484-491
23. Katz F, Malcolm S, Gibbons B et aJ (1988) Cellular and molecular
studies on inf"mt null acute lymphoblastic leukemia. Blood
71.1438-1447
24. Kurzrock R, Blick MB, Talpaz Met al. (1986) Rearrangement in
the breakpoint cluster region and the clinical course in Phifadelphia-negative
chronic myelogenous leukemia. Ann Intern Med 105.673 679
25. Lampert F, Harbott 1, Ritterbach 1 et al. (1988) T -cell acute
childhood lymphoblastic leukcmia with chromosome 14qll anomaly.
a morphologic, immunologic, and cytogenetic analysis or 10 patients.
BJut 56.117-123
26. Lauer SJ, Pinkel D, Buchanan G R et al. (1987) Cytosine arabinosidc/cyclophosphamide
pulses during continuation therapy for childhood acutc lymphoblastic
leukemia. Cancer 60.2366-2371
27 Lipshultz SF, Colan SD, Sanders SP ct al
(1987) Latc cardiac effects of doxorubicin in childhood acute lymphoblastic
Ieukemia (J\LL) (Abstr). Proceedings of the Amcrican Society of
Hematology, 234a
28. Luster AD, Jhanwar SC, Chaganti RSK ct al. (1987) Interferon-inducible
gene maps to a chromosomal band associated with a( 4; II) translocation
in acute
leukemia cells. Proc Natl Acad Sci, USA 84:2868-2871
29. Miller RW, McKay FW (1984) Dcclinc in US childhood cancer mortality
1950 through 1980. JAMA 251.1567-1570
30. Patte C, Philip T, Rodary C et al. (1986) Improved survival
rate in children with Stage III and IV B cell non-Hodgkin's lymphoma
and Ieukcmia using multiagent chemotherapy. Results or a study of
114 children from the Frcnch Pcdiatric Oncology Society. 1 Clin
Oncol 4.12191226
31. Pedersen- Bjergaard J, Ersboll 1, Hansen VL et al. (1988) Carcinoma
of thc urinary bladder after treatmcnt with cyclophosphamide for
non-Hodgkin's lymphoma. N Engl 1 Med 318: 1028-1032
32. Pinkel D ( 1979) Trcatmcnt of childhood acute lymphocytic leukemia.
Modern Trends in Human Leukemia III. R Neth,RC Gallo, P-H Hofschneidcr
and K Mannwcilcr (cds). pp 25-33. New York
33. Pinkel D (1985) Current issues in the management of children
with acute lymphocytic leukaemia. Postgrad Mcd J 61.93-102
34 Pinkel D (1987) Curing children of Icukcmia. Canccr 59.1683 -1691
35. Pui C-H, Williams DL, KaJwinsky DK et al. ( 1986) Cytogenetic
features and serum lactic dehydrogenase level predict a poor treatment
outcome for children with pre-B-cellleukemia. Blood 67: 1688-1692
36. Raimondi SC, Pui CH, Behm FCJ et al. (1987) 7q32-q36 Translocations
in childhood T ccll Icukcmia. Cytogenetic evidcncc ror involvcmcnt
or thc T cell receptor fi-chain gcne. Blood 69.131134
37 Rivera GK, Mauer AM (1987) Controversies in the management of
childhood acute lymphoblastic leukemia. treatment intensification,
CNS Icukcmia, and prognostic factors. Semin Hcmatol 24: 12-26
38. Row Icy JD (1979) Chromosome abnormalities in leukemia. Modern
Trends in Human Leukemia III. R Neth, RC Gallo, P-H Hofschneider
and K Mannweiller (eds.). pp 43 52. New York
39. Rubin CM, Carrino 11, Dicklcr MN et al. (1988) Heterogcncity
of genomic fusion of BCR and ABL in Philadelphia chromosome-positive
acute lymphoblastic Icukcmia. Proc Natl Acad Sci USA 85.2795-2799
40 Sansone R, Strigini P (1988) Infantile leukcmia with a new chromosomal
rearrangement involving Ilq. Cancer Genet Cytogenet 32.293-294
41. Sandberg J\A (1986) The chromosomes in human leukemia. Semin
Hematol 23.201-217
42. Schabel FM Jr, Skipper HE, Trader MW ct al. (1974) Combination
chemotherapy for spontancous AKR lymphoma. Cancer Chemotherapy Rcports
4: 53- 70
43 Sen L, Borella L (1975) Clinical importancc of Iymphoblasts with
T markers in childhood acute leukemia. N Engl 1 Med 292.828-832
44 Stam K, Hcisterkamp N, Grosveld G ct al. (1985) Evidencc of a
new chimeric bcr/c-abl mRNA in patients with chronic myelocytic
leukemia and the Philadelphia chromosome. N Engl J Mcd 3131429-1433
45. Stass SA, Mirro J Jr (1986) Lineage heterogencity in acute Ieukaemia:
Acute mixed-Iineage Ieukemia and lineage switch. Clin Haematol 15.811-827
46. Steinherz PG, Gaynon P, Miller DR et al. (1986) Improved disease-free
survival of children with acute lymphoblastic leukemia at high risk
for early relapse with the New York regimen A new intensive therapy
protocol: A report from the Childrens Cancer Study Group. J Clin
Oncol 4: 744- 752
47. Sullivan MP, Ramirez I (1982) Contribution of cytosar to T -antigen
positive lymphoid disease control in children given 2nd gencration
LSA2L2 therapy. Proc Am Assoc Cancer Res 23:114
48 Sunami S, Fuse A, Simizu Bet al. (1987) The c-sis gene expression
in cells from a patient with acute megakaryoblasticleukemia and
Down's Syndrome. Blood 70: 368-371
49. Williams DL, Tsiatis A, Brodeur GM et al. (1982) Prognostic
importance of chromosome number in 136 untreated children with acute
lymphoblastic leukemia. Blood 60.864-871
50. Yang-Feng TL, Francke U, Ullrich A (1985) Gene for human insulin
receptor: Localization to site on chromosome 19 involved in pre-B-cell
leukemia. Science 228: 728- 730
51. Zuelzer WW, Flatz G (1960) Acute childhood leukemia. A ten-year
study. Am J Dis Child lOO: 886-907
|