|
Institute of Cytology and Genetics, USSR Siberian Academy of Sciences, Novosibirsk, USSR. Introduction Any genome, except for its presently neutral DNA (i.e. without coding sequence), comprises a perfect ensemble of functional genetic units, the range of these units having its roots in individual exons and being crowned by the most complicated supergene complexes. The whole ensemble is undoubtably a product of mutually adaptive molecular coevolution. Any ecosystem, in turn, is the result of concerted molecular evolution of the species making up the system. At present, when the sequencing of entire genomes is running at a phenomena rate, to construct a theory of molecular coevolution would be of utmost importance both for theoretical molecular biology and genetics and for evolutionary theory itself. All specific forms of adaptive molecular coevolution may be subdivided into intra- and intergenome and into directed and nondirected processes [1]. It is typical of nondirected molecular coevolution that, whatever its mode, mutations occur and are fixed at a rate which, despite their apparent adaptive value, remains on average constant. This fact, however, is at variance with one of the keystone postulates of Kimura 's neutralistic theory. Up until now, only nondirected processes of molecular coevolution have been proposed and studied in details: intergenomically, concerted fix ations of mutant reception and absorption genes in bacteria and phages, respectively [2], different variants of coevolving antigens and antibodies [3], original interactions between natural selection and molecular drive in the coevolution of multiple promoter and enhancer regions in rDNA loci, on the one hand, and the RNA Pol J gene, on the other [4], specific pairs of base substitutions compensating for each other to maintain the rRNA secondary structure [5], etc. All these cases of molecular co-evolution, both intra- and intergenome (in different parasite- host pairs), are in fact variations on a theme, i.e. coevolution. The question of whether coevolution could provide development of multigene systems ab simplecioribus ad complexiora is of especially profound interest. Regarding the genes of the immune system, we have suggested that HIV-Iike viruses could be involved in coevolution of this sort [6]. Coevolutionarily Motivated Complication of Immune Multigene Families Let us consider a hypothetical ancestral organism with a primitive,
poorly differentiated immune system. Suppose the corresponding ancestral
immune cells (prelymphocytes) change their state from L to Tin the
course of ontogenesis, where Land Tare the immature and mature prelymphocytes,
respectively. Suppose also that viruses (V) can only strike the
L-cells, i.e. immature prelymphocytes. We then admit that the molecular-genetic
system of immunity is simple enough for the virus to make use, by
adsorption, of the very receptor of the L-cell that T -cells, in
turn, use to identify and inactivate the virus. The adsorption of
V on L leads to the formation of infected cells (denoted Z), from
which, via lysis, the daughter viral particles come. The T IV binding,
by contrast, leads to the elimination of the viruses. Accordingly,
we can derive the following system of differential equations describing
the dynamics: 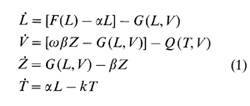
Intragenome Parasites and Genome: a Coevolutionary Aspect We have studied [7] the processes of concerted variability which actually result from cooperation of such entities as, on the one hand, various mobile elements (a kind of "intragenome parasite", GP) and, on the other, the genome itself ("host"). Several systems of differential equations similar to Eq.1 have been built order to analyse the following situations: 1) the GP is insertable in the vacant sites only, its free state (not in the "host" but still in the cell) not being durable; 2) the GP is insertable in the vacant sites only, its free state being durable; 3) the GP is insertable in both vacant and occupied sites ("molecular memory"), its free state not being durable (mammalian Alu-like repeats taken as a prototype ); 4) the GP is insertable in both vacant and occupied sites and is able to exist "on its own" (retroviruses taken as a prototype). We then admitted that the genome is tolerant to the "selfish" proliferation of GP until the share of the occupied sites exceeds the limit 1 I K. Our analysis revealed that the coevolutionary complication of GP from the simplest, which is only able to insert in vacant sites, through the ongoing acquirement of terminal re peats ("molecular memory"), to perfectly integrated complexes with an extragenomiallife style is accompanied by change in the selective coevolutionary restrictions on genome size: upper limit-no limit -lower limit. Thus, mobile elements may be regarded as an inner factor inducing progressive, coevolutionarily motivated complication of genomes, including multiplication of coding regions. Our models are based on the assumption that there is always a superior selective force (from the "host" side) that restricts the number of GPs and influences the pattern of GP distribution in the host genome. However, the following question arises here: Are there any inferior restrictions directly related to the GP structure as such? We go on to show below that A/u-Iike repeated sequences, even with extremely simple structures, could have such restrictions. CpG-Rich Promoters as an Inner Constraint on Amplification of Alu-Like Sequences With the aid of the package of applied programs VOSTORG [8], designed
in our laboratory, 83 A/u repeats (60 human included) from seven
species of primates and 13 A/u-Iike B1 repeats from three rodent
species were subjected to phy logenetic analysis, in particular,
for mutations fixed in RNA polymerase III promoters (Fig. 1 ). Using
the method of diagnostic positions [9] enabled us to divide all
60 human A/u sequences into three different classes (Fig. 2) corresponding
to J, Sa and Sb (identification of the Sc class was certain) according
to Brit ten et al. [9]. The topologies of the phylogenetic tree
constructed on the complete sample of A/u sequences and of the tree
derived from the comparison of the consensus for all classes revealed
a good agreement with the order of appearance of these classes in
the course of evolution (Fig. 3): progenitor (7 SL RNA gene) --.J--.
Sa --. Sc(?) --.Sb. As is known, the CpG positions evolve on average
10.5 times as much as other positions of A/u repeats, which is due
to methylation of the cytosines. In particular, the A (enhancing)
and B (initiating) boxes of promoters contribute considerably to
the concentration of CpGs (Fig. 1). We tried to build a dichotomic
dendrogram from CpG positions of the promoter alone but failed.
This could be an argument in favour of the "burst"-like formation
of the A/u classes. The most intriguing feature of the A/u evolutionary
tree (Fig. 3 c) is the almost absolute lack of mutations in CpG
dinucleotides of the promoter region at the 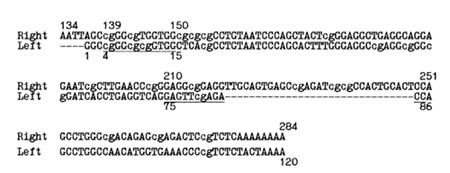 Fig. 1. Consensus of human Alu repeats [9] with the left and right halves of the sequences aligned. A and B boxes of the promoter region are underlined. The CpG dinucleotides are in lower case letters. The right promoter is likely to be inactive due to the relatively long inserted sequence 
upper branches of the tree (when the Alu subfamilies wcre being
newly formed), in spite of their extreme mutability. NonCpG positions
show the same regularity (Fig. 3 e). It should be noted that mutations
in the CpG sites of the "quasineutral" part of the Alu repeats appear
not to be in deficit at that period (Fig. 3 d). Thus, a lion's share
of mutations in the promoter CpG sites are concentrated in the lower
branches of the divergency tree just after the class formation process
is over (not shown). This means that the promoter region of Alu
repeat progenitors were under very strong negative natural selection
pressure until the amplification process started. Moreover, the
topology of the dichotomic branching within each class appears to
be unstable. Thus, during evolution, first some changes in diagnostic
positions (CpG sites not belonging to them) had to be accumulated;
secondly, a current class of Alu sequences branched off thc main
stem of the trcc; and, finally, mutations at CpG positions predominantly
within the promoter occurred most rapidly. The superfamily of Bl
repeats of rodents, closely related to Alu, shows similar regularities.
The results obtained allow us to propose a model where promoter
sites playa role of profound importance both in intragenome amplification
of the progenitor Alu sequence and in the divergence of individual
members of the corresponding subfamilies. The model is supposed
to explain the limited sizes of a subfamily with the subsequent
acquisition ofmutational defects in CpG positions of promoters and
hence the inevitable slowdown of amplification. As a result, only
those of 7 SL RNA-Iike sequences which have retained the promoters
could 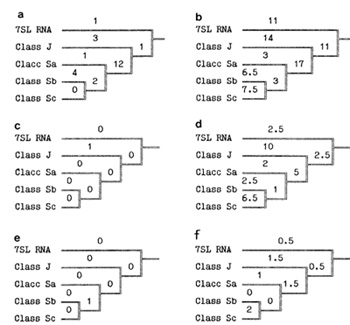
become the progenitor for the following subfamily of Alu repeats to amplify and evolve in an active mode. Each Alu repeat is well known to consist of two homologous halves (Fig. 1 ). Usually , only the leftmost domain is active for amplification by reverse transcription [10]. Figure 3f shows that, in contrast to the single CpG mutation in the leftmost promoter, the rightmost one accepted seven such mutations in CpG dinucleotides at the top part of the tree just when the Alu subfamilies were in the making. This is an additional, rather convincing, argument in favour of the importance of the promoter CpG sites, in particular those located in A and B boxes. Thus, the "selfish" intragenome propagation of any progenitor "pregnant" with a recurrent Alu subfamily is destined to slow down and, eventually, to come to a standstill because as any individual Alu promoter rapidly accumulates more and more defects, predominantly due to the increased mutability of CpG sites, the host reverse transcriptase becomes less able to recognize the promoter. This is not so with HIV-Iike retroviruses. They show unusually high variability, generated by viral reverse transcriptase, the most error-prone of the various RNA and DNA polymerases [11 ]. In contrast to the short Alu repeating unit, the HIV reverse transcriptase is encoded by its own Pol gene. It produces 327 extremely frequent mutations in all regions of the viral genome, including in its own gene. Therefore, there is a good chance for promoter sites and reverse transcriptase to be involved in prolonged steady coevolution, based on the selection of pairs of substitutions compensating for each other. It is an original case of a strikingly rapid intragenome coevolution which should be adaptive but is apparently not directed. Summary and Conclusions "Parasitic" DNA may be regarded as a rather active partner in different coevolutionary processes. The basic stages of the processes are likely in most cases to be as follows. parasitism? tolerance -+ -+ symbiosis. There are interior and exterior coevolutionary factors complicating molecular-genetic systems within a supersystem "mobile elements-genome". For example, the data presented above indicate clearly that the relatively high concentration of CpG sites in the Alu promoter looks prudent as regards the needs of the "parasite" as well as those of the host genome. We consider "prudence" of this kind to be most likely a product of large-scale molecular coevolution. As to HIV -like retroviruses, they could be simultaneously involved in three different regimes of molecular coevolution: 1) at a level of the parasitic genome as such; 2) as atypical intragenome parasite inserted in the host genome inducing complication in multigenic system (like Alu); 3) as a typical intracellular parasite in an "active", infectious state stimulating complication in the immune multigene families. Evidently, it is only the steadiness of the first coevolutionary process (with "no wheels, no sails") provides for a possible 328 role of HIV-Iike parasites as a selective factor provoking coevolutionary complication of host genomes. References 1. Rodin SN, Rzhetsky AY, Matushkin YG (1987) Coevolutionary approach
to the motivation of molecular-genetic organization of immune system.
In. MlikovskyJ, Novak V (eds) Towards a new synthesis in evolutionary
biology. CSA V, Prague, pp130-132 |