|
Departments of Medical Genetics and Pediatrics University of Wisconsin Madison, WI 53706 USA
Melanocytes, which constitute only a minor population in the skin and eyes of mammals, are the only cells that produce melanin, which accounts for virtually all of the visible pigmentation of those tissues Skin melanocytes are highly dendritic cells that originate during development as melanoblasts in the neural crest, whereas melanocytes in the retina derive from the optic cup The melanoblasts subsequently migrate to the epidermal/dermal border of the skin, the hair bulbs in the dermis of the skin, and the iris, choroid, and retina of the eye (reviewed in Spritz and Hearing, 1995) For pigmentation to proceed normally, melanoblasts must thus differentiate appropriately as melanoblasts in the neural crest, the signal for migration must be given at the correct time, and movement of melanoblasts towards their eventual destination must begin To achieve uniform pigmentation, melanoblasts must not only disperse correctly, but must subsequently receive the appropriate signal to stop migrating, establish themselves, proliferate and differentiate appropriately, and of course function correctly. The signals necessary to achieve all of these events necessarily depend on a number of genes, some expressed by the melanoblasts and/or melanocytes while others are expressed by other cell types Since the ultimate patterns of pigmentation in the skin and hair depend on other cellular constituents, such as keratinocytes, genes which regulate those other cell types can also indirectly impact on pigmentation More than 150 different mutations that affect pigmentation have been identified in the mouse, and these map to more than 60 distinct genetic loci (Silvers, 1979; Lyon and Searle, 1989) A number of these genes have now been cloned and will be discussed in the relevant sections that follow Within the melanocyte, the pigment biosynthetic machinery is confined to membranebound organelles termed melanosomes Melanin is deposited on the internal fibers of the melanosome, and as melanization proceeds the melanosomes are moved steadily away from the perinuclear region down the dendritic processes of the melanocyte, and are eventually transported to keratinocytes, where they are subsequently degraded. Melanin biosynthesis initiates from the amino acid tyrosine, and ill vitra only a single enzyme is absolutely necessary to synthesize melanin tyrosinase (monophenol mono oxygenase; monophenol, L-dopaoxygen oxidoreductase; EC 1.14.18.1) Tyrosinase is a copper-containing enzyme that catalyzes the first two steps of melanin biosynthesis, the hydroxylation of tyrosine to 3, 4-dihydroxyphenylalanine (DOP A) and the subsequent oxidation of DOP A to DOP Aquinone The first reaction is the most critical since its spontaneous rate is negligible, whereas DOP A can readily auto oxidize to DOP Aquinone even in the absence of tyrosinase. DOP Aquinone then continues through the melanin biosynthetic pathway via a series of reactions will take place spontaneously in a test tube, but ill viva involve several other enzymes, including DOP Achrome tautomerase and DHICA oxidase These enzymes have been characterized biochemically, and the corresponding genes have been cloned and are discussed in detail below. Two different basic types of melanin can be produced in mammalian melanocytes, brown/black eumelanin and yellow/red pheomelanin Relatively little is known about the controls that modulate biosynthesis of eumelanin versus pheomelanin. Melanin was initially thought to be an homogeneous high molecular weight biopolymer consisting exclusively of an indole-5,6-quinone backbone, but physiological melanins are now known to be much more heterogeneous, incorporating incorporate other intermediates in the melanin pathway as well
ALBINISM Albinism is a heterogeneous group of disorders of melanocyte function, characterized principally by generalized hypopigmentation In oculocutaneous albinism (OCA) pigment is reduced or absent in the skin, hair, and eyes, whereas in so-called "ocular albinism" (OA) visual involvement is accompanied by relatively normal pigmentation of the skin and hair In fact, however, all forms of human albinism are oculocutaneous, involving both the eyes and the integument to at least some extent; thus, the distinction between OCA and OA is largely artificial The role played by melanin in the developing visual system is not known. However, if retinal pigment production is reduced below some critical level, regardless of its genetic cause, there results a stereotypic set of defects of neuronal migration in the visual pathways, with consequent low vision, nystagmus, and strabismus in affected individuals In general, the severity of the visual defects correlates with the severity of the pigmentation deficit in the eye OCA has traditionally been classified as "tyrosinase-deficient" or "tyrosinase-positive" on the basis of biochemical assay of hairbulb tyrosinase, corresponding approximately to the current genetic designations, OCA1 and OCA2, although several additional tyrosinase-positive types of OCA have been distinguished on clinical grounds Both X-Iinked and autosomal recessive forms of OA have been recognized However, elucidation of the molecular basis of these disorders has led to improved understanding of their true relationships and to more accurate classification on molecular genetic grounds.
OCA1 is an autosomal recessive disorder resulting from deficient
catalytic activity of tyrosinase, the enzyme that catalyzes the
first two steps, and at least one subsequent step, in the melanin
biosynthetic pathway (reviewed in Spritz and Hearing, 1995) As discussed
above, tyrosinase is absolutely necessary for pigment production,
although other control points in the pathway also regulate melanogenesis
in viva Freshly epilated hairbulbs from patients with type OCA1
accumulate little or no melanin pigment on incubation in tyrosine
or dopa i"n vifro, the traditional assay for this disorder (Kugelman
and van Scott, 1961; reviewed in Witkop, 1989); however, this test
is highly unreliable and is therefore not useful clinically. In
humans at least two different subtypes of OCA1, OCA1A and OCA1B,
have been distinguished on clinical grounds In OCA1A ("tyrosinase-negative
OCA"), the classic, most severe form of the disorder, tyrosinase
activity is completely absent, and there is no detectable melanin
pigment in the skin, hair, or eyes Visual acuity is greatly decreased,
usually to approximately 20/400, and nystagmus, strabismus, and
photophobia are usually severe. In OCA1B ("yellow OCA"), initially
recognized in the Amish (Nance et al, 1970), tyrosinase activity
is greatly reduced, and there is little or no apparent melanin pigmentation
at birth, although progressive melanization may occur during childhood
and adult life OCA1A and OCA1B have long been recognized to be allelic,
based on the existence of apparent OCA 1 A/OCA 1 B compound heterozygotes
who exhibit a phenotype approximately intermediate between those
of the two homozygous forms (Hu et al, 1980) Analyses of the human
tyrosinase structural gene ( TYR) in patients with OCA 1 have demonstrated
that an allelic series of tyrosinase gene mutations accounts for
both OCA 1 A and OCA1B The gene consists of five exons spanning
more than 65 kbp ofDNA on the long arm of human chromosome II (Giebel
et al, 1991a) Tyrosinase is translated as a 529-amino acid polypeptide,
from which an 18-amino acid hydrophobic leader peptide is cleaved
to yield the 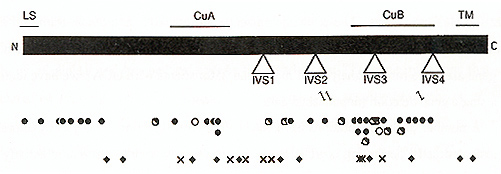
mature tyrosinase (Kwon et al, 1987; Wittbjer et al, 1989) An apparent transmembrane domain near the carboxyl terminus of the polypeptide indicates that tyrosinase is membrane bound, most likely on the inner surface of the melanosome Among Caucasians, two codons, 192 and 402, are commonly polymorphic (Giebel and Spritz, 1990; Tripathi et al, 1991), encoding either of two alternative amino acid residues at each site. As a result, there are four slightly different alternative normal tyrosinase isoforms, which differ significantly in their stabilities and thus also in their net catalytic activities (Tripathi et al., 1991). However, the occurrence of these four tyrosinase isoforms does not obviously correlate with the apparent pigmentation phenotypes among normal Caucasians OCAI was genetically linked to the TYR gene by RFLP analysis of large families with OCA (Giebel et al., 1990; Spritz et al, 1990), and subsequently specific TYR gene mutations were demonstrated by DNA sequence analyses of TYR cDNAs cloned from cultured melanocytes (Tomita et al 1989) and TYR genomic segments amplified from patient DNAs by the PCR (Giebel et al, 1990; Spritz et al, 1990) As shown in Figure 1, at least 61 different pathologic mutations of the TYR~ gene have been identified to date (reviewed in Spritz, 1994a; Spritz and Hearing, 1995) These include missense and nonsense mutations, frameshifts, and mutations that interfere with normal pre-mRNA splicing Surprisingly, deletions of the human TYR gene are quite rare; in analyses of more than 250 patients with OCA1 we have identified only a single gene deletion (unpublished data) A number of these mutations have been subjected to functional analysis by expression in transfected cells, permitting correlation of the resultant tyrosinase enzymatic activity with the associated clinical phenotypes (reviewed in Spritz and Hearing, 1995). As expected, the nonsense, frameshift, and the missense mutations that completely abolish tyrosinase catalytic activity are associated with OCA1A. However, missense mutations that result in greatly reduced, but not abolished, enzymatic activity are specifically associated with OCA1B. In particular, the V275F, P406L, and R422Q missense substitutions, and also the R402Q polymorphism, result in unstable tyrosinase polypeptides, with temperature-sensitive catalytic activities that are greatly reduced at 37oC compared to 31oC, both in vivo and in vitro. As illustrated in Fig. 1, the nonsense and frameshift mutations are randomly distributed throughout the tyrosinase coding region However, most of the missense substitutions associated with OCA1A cluster in two regions of the polypeptide (King et al., 1991; Tripathi etal., 1992): nearthe amino terminus of themature tyrosinase polypeptide (codons21-89) and near the center of the tyrosinase polypeptide (codons 371-448). This latter region corresponds closely to the so-called "Cu B" region of tyrosinase. This is one of two regions of the tyrosinase polypeptide that exhibit amino acid sequence homology to arachnid, crustacean, and molluscan hemocyanins (Lerch, 1988), proteins which, like tyrosinase, also complex with atomic copper. However, involvement of the Cu B region in copper binding by tyrosinase has yet to be demonstrated experimentally, and the only amino acid substitution we have found that involves one of the putative copper-binding histidines (Martinez et al., 1985) (H390D) is associated with a particularly mild OCA phenotype (unpublished data). In Caucasians, no single mutant TYR allele accounts for a significant fraction of the total (reviewed in Spritz, 1994a; Spritz and Hearing, 1995); therefore, in the absence of parental consanguinity most patients are compound heterozygotes for different mutant alleles Patients with OCAIA have two OCA1A alleles, whereas patients with OCA1B may have either two OCA1B alleles or one OCA1B and one OCA1A allele. However, the range of phenotypes associated with OCA1B mutations is very broad, even among patients with similar genotypes, probably because of epistatic phenomena that modulate pigmentation in the presence of even a small amount of tyrosinase activity In fact, we have found that approximately 25 percent of Caucasian patients diagnosed with "tyrosinase-positive OCA" on the basis of clinical and biochemical criteria have pathologic TYR gene mutations that would be expected to only modestly reduce tyrosinase catalytic activity (Tripathi et al, 1992; unpublished data) Furthermore, we recently identified mutations of the TYR gene in two patients with apparent "autosomal recessive ocular albinism" (AROA; MIM #203310) (Fukai et al., 1995) Interestingly, both of these patients are compound heterozygotes for OCA1A mutant alleles and the R402Q polymorphic allele, which is associated with moderate thermoinstability of the corresponding tyrosinase polypeptide; thus these patients actually have very mild forms of OCA1B. Similarly, other cases of AROA appear to represent mild forms of OCA2 (see below). Thus, AROA does not appear to represent a distinct genetic entity. These observations underscore the need for molecular diagnosis to accurately distinguish these disorders. We have studied several examples of OCA1 occurring in two successive generations ofa kindred (Giebel et al., 1990, 1991b; unpublished data) All of these cases were found to result from pseudodominance, an affected individual having inadvertently married an unrelated carrier. It would be of great interest to determine whether this phenomenon might account for some or all of the reported cases of "autosomal dominant OCA", which has previously been considered to be a distinct entity (MIM #126070). The lack of predominant mutant TYR alleles among Caucasian patients with OCA1 complicates efforts at molecular diagnosis or carrier detection, except in selected families However, in certain ethnic subgroups one specific mutant allele strongly predominates (reviewed in Spritz, 1994a; Spritz and Hearing, 1995) In these populations, therefore, genetic screening procedures can be targeted for the appropriate predominant mutant TYR alleles. We have carried out DNA-based carrier detection of OCA1 in a number of instances, and recently carried out DNA-based prenatal diagnosis of OCA 1 A in an Israeli family (FalikBorenstein et al., 1995)
OCA2 is an autosomal recessive disorder with a phenotype similar
to, but usually less severe than, OCA 1, although the two disorders
display considerable clinical overlap Infants with OCA2 may have
little or no apparent melanin pigment at birth. However, during
early to mid childhood, most patients with OCA2 acquire modest amounts
of pigment, often predominantly yellow-red pheomelanins The visual
deficits are also usually less severe in OCA2 than in OCA 1, the
visual acuity of patients with OCA2 is typically in the 20/60 to
20/200 range, with only moderate nystagmus Furthermore, in OCA2
visual acuity and nystagmus may improve during childhood and even
during adolescence, with ultimate visual acuity reaching approximately
20/40 to 20/70 Rarely, nystagmus may even disappear entirely and
visual acuity may become normal (Summers et al, 1991) As described
above, molecular analysis has shown that a significant fraction
of Caucasian patients thought to have "tyrosinase-positive" OCA
actually have clinically mild forms of OCA1B However, the existence
ofa true OCA2, genetically distinct from OCA1, has long been known
on the basis of several instances of matings between parents, one
with OCA1 and the other with OCA2, that produced normally pigmented
children (Witkop et al, 1989) As will be discussed below, OCA2 is
only one of perhaps several different types of "tyrosinase-positive
OCA", although it is likely the most frequent type, especially in
African and African-American populations Recently, we identified
the gene responsible for OCA2 in humans and we have characterized
mutations of this gene in a number of patients with OCA2 (Rinchik
et al, 1993; Lee et al, 1994a,b) The human type II OCA gene, p (Rinchik
et al, 1993) proved to correspond to the pink-eyed dilution p) locus
of mouse (Gardner et al., 1992; Rinchik et al 1993), which is associated
with a mutant phenotype very similar to that of human OCA2 (reviewed
in Silvers, 1979; Lyon and Searle, 1989). The human p gene is located
in chromosome segment 15q 11-q 13, in agreement with mapping of
the human OCA2 locus by genetic linkage (Ram say et al, 1992) Furthermore,
the p gene is located adjacent to the chromosomal region commonly
deleted in patients with the Prader-Willi (PWS; MIM #176270) and
Angelman (AS; MIM #105830) syndromes (reviewed in Nicholls, 1993),
both of which are often associated with significant hypopigmentation
(Butler, 1989) Approximately one percent of patients with PWS and
AS also have OCA2, similar to the expected prevalence of OCA2 heterozygotes,
suggesting that OCA2 in association with PWS and AS might result
from hemizygosity (or, rarely, uniparental isodisomy) for inherited
p gene mutations on the nondeleted chromosome, and that isolated
OCA2 might result from point mutations of the p gene 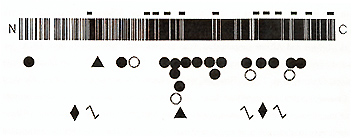
To test these hypotheses, we first studied one such patient with both PWS and OCA2, in whom we found a large de novo deletion of proximal 15q (including P) on the paternal chromosome, accounting for PWS, and a hemizygous maternally inherited mutant p allele containing a small partial gene deletion, accounting for OCA (Rinchik et al, 1993) Subsequently, to identify point mutations in patients with isolated OCA2, we characterized the p genomic locus, which consists of25 exons spanning 250-600 kbp ofDNA on proximal 15q (Lee et al., 1995). Using PCR primers based on these data, we have identified 25 additional different abnormalities of the p gene in other patients with OCA2, including two more patients with both OCA2 and PWS (Lee et al, 1994a,b) As shown in Fig 2, these include large intragenic deletions, small in-frame deletions, frameshifts, splice junction mutations, and missense substitutions In Caucasian and African-American patients none of these mutations are strongly predominant, although one substitution, V443I, is relatively frequent in both groups (Lee et al., 1994a,b; unpublished data). However, we found that one mutation, a 2.7kb deletion encompassing only P exon 7 (Durham-Pierre et al., 1994; Lee et al, 1994b ), is strongly predominant in Tanzanian patients with OCA2 (unpublished data) OCA2 is very frequent in this group, occurring with an incidence of approximately 1 per 1300 (Luande et al, 1985), and OCA2-associated skin cancers constitute a major public health problem in Tanzania. It thus seems likely that the repertoire of OCA2 mutant alleles in east Africa differs from that in west Africa, the population from which most African-Americans are derived Interestingly, we have found p gene mutations in all twenty unrelated African-American (Lee et al, 1994a) and African (unpublished data) patients with "tyrosinase-positive OCA" we have studied, consistent with complete linkage of OCA2 to 15q markers in South African patients (Ramsay et al, 1992) However, we have found p gene mutations in about one-third of Caucasian patients (unpublished data) As discussed above, about 25% of these patients have mutations in the TYR gene, and, as will be discussed below, at least some of these patients may have mutations of a presumed third OCA locus The reason for this unexpected difference between African-American and African versus Caucasian patients with "tyrosinasepositive OCA" is not known, but may be the result of historically different selection pressures on these two populations The range of clinical phenotypes associated with pathologic mutations of the p gene in humans is very broad Individuals homozygous for mutations that would be expected to totally abolish p function exhibit a phenotype almost as severe as OCA 1 A (Lee et al. 1994b; unpublished data). At the other end of the range, mutations of p that would be expected to only partially reduce function appear to account for some cases of AROA (MIM #203310) (Lee et al., 1994a,b). AROA thus is heterogeneous in nature, in different patients from mild mutations of the TYR, P, and perhaps other genes involved in pigmentation Although the p gene product has not yet been characterized experimentally, it is expected to play an important role in eumelanotic pigmentation. In mouse, p mRNA is only expressed in viva in melanocytes that synthesize brown-black eumelanin; melanocytes that specifically synthesize pheomelanin do not express p (Rinchik et al., 1993). The predicted human p polypeptide includes a total 12 transmembrane domains, in an arrangement characteristic of proteins that transport small molecules such as amino acids. This suggests that the p protein may be an integral component of the melanosomal membrane, possibly involved in the transport of tyrosine, the immediate precursor to melanin biosynthesis. This would be consistent with the observation that pigmentation of melanocytes from humans with OCA2 (reviewed in Witkop et al, 1989) and from homozygous p mutant mice (reviewed in Silvers, 1979) is greatly increased by incubation with excess exogenous tyrosine More important, this suggests that treatment with exogenous tyrosine or other agents may offer an approach to pharmacologic therapy for patients with OCA2
Originally described in patients from Nigeria, "brown OCA" is associated with moderate hypopigmentation of the skin, hair, and eyes, and modest ocular dysfunction Brown OCA has long been considered a possible human homologue of brown (b) mutant mice, which exhibit brown coat color (Silvers, 1979; Lyon and Searle, 1989) Patients with so-called "brown OCA" are tyrosinase-positive, and are not clinically distinguishable from patients with milder forms of OCA2 Thus, the issue of allelism between human "brown OCA" and OCA2 remains open Recently, however, some evidence that abnormalities of the human b locus ( TYRP; MIM # 11550 I) might be associated with hypopigmentation in some patients has begun to emerge A cDNA for mouse b was the first member of the tyrosinase gene family to be (inadvertently) cloned (Shibahara et al., 1986), and was quickly shown represent the b locus, rather than tyrosinase itself (Jackson, 1988; Bennett et al., 1990). The amino acid sequence of the b gene product is very similar to that of tyrosinase, and hence was named "tyrosinaserelated protein" (TRP-I ); it corresponds to a melanosomal protein previously termed "gp75 (Vijayasaradhi et al, 1990) In the mouse, b locus mutations result in the production of brown rather than black eumelanin, and recent biochemical studies indicate that TRP-1 corresponds to 5,6-dihydroxyindole-2-carboxylic acid (DHICA) oxidase (Jiminez-Cervantes et al., 1994; Winder et al., 1994), an enzyme catalyzing a distal step in the eumelanin biosynthetic pathway. The homologous human cDNA has been cloned (Cohen et al., 1990) and the gene (TYRP) mapped to chromosome segment 9p22-p23 (Chintamaneni et al, 1991; Murty et al., 1992), and Wagstaff(1993) demonstrated that TYRP is deleted in two patients with the 9psyndrome associated with moderate hypopigmentation. Furthermore, Boissy and coworkers (1993) found virtually no immunologically detectable TRP-1 protein or mRNA in one patient with "brown OCA", and recently (Wildenberg et al., 1994) identified a frameshift mutation of the TYRP gene in this patient, who thus has "OCA3". However, the hypopigmentation phenotype of this patient is rather less severe than that of most patients with "tyrosinasepositive OCA", and it remains to be seen what fraction of such patients have mutations of TYRP.
Ocular albinism of the Nettleship-Falls type (OAI) is an X-Iinked recessive disorder clinically similar to AROA In affected males mild cutaneous hypopigmentation is accompanied by hypopigmentation of the iris and retina and foveal hypoplasia, resulting in reduced visual acuity, photophobia, nystagmus, and strabismus In some, but not all affected individuals, electron microscopy of skin demonstrates melanin macroglobules, often termed "macromelanosomes". Carrier females may exhibit a heterogeneous distribution of retinal pigmentation, sometimes referred to as a "tigroid" or "mud-spattered" pattern, which is thought to be indicative ofX-inactivation Efforts at positional cloning of the OA 1 gene are far advanced. The gene was assigned to chromosome segment Xp223 by genetic linkage, with localization most likely between markers DXSI43 and DXS85 (Bergen et al, 1990, 1991; Schnur et al, 1991; Charles et al, 1992). Sunohara et al. ( 1986) described a patient with three apparent X-Iinked recessive disorders; OAI, X-Iinked ichthyosis, and Kallmann syndrome, indicating that these three genes may be in close physical proximity and be deleted in this patient (Andria et al., 1987) Furthermore, the colocalization of both OAI and "X-Iinked ocular albinism and deafness" (OASD; MIM #300650) to Xp223 suggests that OASD may also result from a microdeletion that includes the OA 1 gene (Winship et al., 1993) By detailed mapping of the X chromosomal deletions in the patient reported by Sunohara et al ( 1986) and a second, similar patient, Wapennar et al. (1993) localized the OAl locus to a 200-kbp interval between markers 210B5-R and A187H6-41A They assembled a 2.6-mbp YAC contig spanning this region, part of a larger contig spanning chromosome segment Xp22 (Schaefer et al., 1993). It seems likely that these Y ACs will be useful reagents for the eventual isolation of the OAl gene Interestingly, the OA 1 locus is located within 1-2 Mb of the gene for "microphthalmia with linear skin defects" (MLS; MIM #309801) (Wapenaar et al, 1993) Although there is not a clear homologue to human OAI in the mouse, synteny between the human and mouse Xchromosomal maps suggests that the mouse "lined" mutation (Li) might be a chromosomal deletion encompassing MLS and possibly also OAI (Lyon and Searle, 1989)
Several additional human autosomal recessive genetic disorders, including Chediak-Higashi syndrome, Hermansky-Pudlak syndrome, and Cross syndrome, to name but three, are characterized by complex phenotypes that include moderate to severe manifestations of OCA The genes responsible for these disorders have not yet been identified Even more intriguing, we have recently obtained evidence for the existence of at least one additional human OCA locus As discussed above, many patients with "tyrosinase-positive OCA" genes do not have identifiable mutations of either the TYR or p genes Recently, we studied one of these large families by genetic linkage analysis, and excluded abnormalities of TYR, P, and also TYRP (unpublished data). This indicates that tyrosinase-positive OCA in at least this kindred results from abnormalities of a different, as-yet unknown, locus Identification and analysis of this and perhaps other human OCA genes will thus constitute major challenges as our understanding of these most ancient human genetic disorders continues to progress.
Piebaldism and Waardenburg syndromes are distinct inherited disorders characterized in part by striking areas of congenital depigmentation These disorders contrast with albinism, in which relatively homogeneous hypopigmentation results from defects of melanocyte function, and with vitiligo, in which acquired areas of depigmentation result from reduced melanocyte survival In piebaldism the abnormal distribution of pigment results from abnormal distribution of melanocytes during development, most likely due to defective proliferation of melanoblasts prior to migration to the dermis during embryogenesis. In piebaldism the pigmentary anomalies constitute the principal clinical manifestation, whereas in Waardenburg syndrome deafness and dystopia canthorum (lateral displacement of the eyes) are also of major clinical importance The elucidation of the molecular defects underlying piebaldism and Waardenburg syndrome provide additional examples of the use of human and mouse genetics in combination to dissect the molecular basis of genetic diseases
Piebaldism is an autosomal dominant disorder of melanocyte development
characterized clinically by congenital patches of white skin (leukoderma)
and white hair (poliosis), principally located on the scalp, forehead,
ventral chest and abdomen, and extremities (Keeler, 1934; Froggat,
1951; Cooke, 1952) In contrast to vitiligo, with which it is frequently
confused, in piebaldism the depigmented patches are present from
birth and are generally static in shape and distribution, although
limited filling in sometimes occurs, especially in milder cases
Hirschprung disease is occasionally also present, but most affected
individuals display only the pigmentary anomaly Piebaldism is relatively
rare, although its actual frequency is not known Because of its
distinctive phenotype, piebaldism, sometimes incorrectly called
"partial albinism", was one of the first autosomal dominant genetic
disorders recognized (Morgan, 1786; Lucian, 1905), and was one of
the first genetic disorders for which a pedigree was presented (reviewed
in Froggat, 1951) Histologically, the depigmented patches lack most
or all melanocytes (Breathnach et al., 1965; Jimbow et al, 1975),
although areas of increased pigmentation may occur at the boundaries
of, or even within, the regions of hypopigmentation Melanocytes
in the skin and hairbulbs derive embryologically from the neural
crest, and piebaldism has thus been considered to be a lineage-specific
disorder of neural crest development, most likely involving defective
melanoblast proliferation, migration, or survival (Murphy et al.
, 1992; Steel et al. , 1992) The critical clues to the molecular
basis of human piebaldism came from studies of a similar disorder
of mice, "dominant white spotting" (W) (reviewed in Spritz, 1993,
1994b) Mouse dominant white spotting, which is associated with defects
of pigmentation, hematopoiesis, and germ-cell development (reviewed
in Silvers, 1979; Lyon and Searle, 1989), was found to result from
deletions or point mutations of the c-kit protooncogene (Chabot
et al. , 1988; Geissler et al. , 1988; Reith et al. , 1990; Tan
et al. , 1990; reviewed in Morrison-Graham and Takahashi, 1993)
C-kit, originally identified in the genome of the HZ4-feline sarcoma
virus (Besmer et al , 1986), encodes the cell-surface receptor for
an embryonic growth factor, variously called "steel factor" (SLF),
"mast cell growth factor", "stem cell factor", "KL" (kit ligand)
in the literature (Copeland et al, 1990; Flanagan and Leder, 1990;
Huang et al, 1990; Zsebo et al, 1990; Murphy et al, 1992; reviewed
in Morrison-Graham and Takahashi, 1993) The KIT receptor is expressed
by melanocytes, whereas SLF is expressed by other, as yet undefined,
cell types SLF is the product of the steel (SI) locus of mice, and
both SI and W mutant mice display similar piebald-like phenotypes
However, SI mutant melanocytes develop normally when transplanted
to normal skin (since normal ligand is available) whereas W mutant
melanocytes (with an inherently defective receptor) 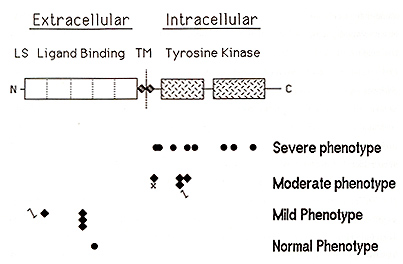
do not The human piebaldism locus was mapped to chromosome segment 4q 12 on the basis of several patients with piebaldism and de nOVO chromosomal translocations or deletions involving this region (Funderburk and Crandall, 1974; Lacassie et al., 1977; Hoo et al., 1986; Yamamoto et al., 1989). Subsequently, the human KITgene was mapped to approximately the same chromosomal location (Yarden et al., 1987; d'Auriol et al, 1988), suggesting that human piebaldism might, like mouse "dominant white spotting", also result from abnormalities of the KIT gene The KIT protein is a member of the tyrosine kinase family of transmembrane receptors As illustrated in Figure 3, the KIT polypeptide consists of an amino-terminal extracellular ligand-binding receptor domain composed of five immunoglobulin-type repeats, a short transmembrane domain, and an intracellular domain consisting of a bipartite tyrosine kinase domain followed by a carboxyl-terminal tail The stoichiometry of interaction between SLF and the extracellular domain of the KIT receptor is not certain, but is thought to be monovalent (Lev et al, 1992) On binding SLF, the KIT receptor dimerizes within the cell membrane (Level al. , 1992), activating its intracellular tyrosine kinase This, in turn, results in autophosphorylation of specific tyrosine residues within the KIT kinase domain, enhancing the binding of various proteins, including phosphatidylinositol 3' kinase and phospholipase Cy 1, which act as downstream mediators of the mitogenic signal in the KIT -dependent pathway of signal transduction (reviewed in Morrison-Graham and Takahashi 1993), In the mouse, KIT function is required both immediately prior to melanoblast migration and also postnatally (Nishikawa et al. , 1991 ), and we have shown that KIT function is required for proliferation, although not survival, of human melanocytes in culture (Spritz et al, 1994a) It seems likely, therefore, white spotting in both human piebaldism and mouse W results deficient proliferation of melanoblasts prior to migration during development The human KIT gene consists of 21 exons spanning more than 70 kb at chromosome segment 4q 12 ( Giebel et al. , 1992; Vandenbark et al. , 1992), part of a cluster of genes encoding type III receptor tyrosine kinases, organized PDGFRA-KIT-KDR (Spritz et al., 1994b) The first direct evidence for the involvement of KIT mutations in human piebaldism came from Giebel and Spritz ( 1991 ), who studied an extended kindred with piebaldism and identified a missense mutation, G664R, within the highly conserved intracellular tyrosine kinase domain, and demonstrated linkage with a lod score in excess of 6 with no recombinants Further support came from analyses of two additional patients, one with a cytogenetic deletion of 4q 12-q211 (Yamamoto et al. , 1989), in both of whom KIT and PDGFRA were found to be deleted (Fleischman et al., 1991; Spritz et al., 1992), As shown in Figure 3, a total of 18 different point mutations of the KIT gene have now been identified in different families with piebaldism Interestingly, as also shown in Figure 3, these mutations can be classified into four general groups, each group tending to be associated with piebald phenotypes of differing severity Thus, a hierarchical paradigm of pathologic human Kll' mutations appears to account for a graded series of dominant phenotypes in human piebaldism The first group of KIT gene mutations consists of missense substitutions, all but one of which have been located within the highly conserved tyrosine kinase domain and almost all of which involve amino acid residues that have been particularly conserved Several of these human KIT. substitutions correspond closely to the positions of similar mutations in various strains of W mutant mice (reviewed in Morrison-Graham and Takahashi, 1993), underscoring the importance of these sites to function of the KIT receptor In fact, the human E583K mutation corresponds precisely to the mouse W37 mutation (Fleischman, 1992) All of the KIT missense substitutions in the kinase domain are associated with relatively severe piebald phenotypes This is a consequence of the fact that the KIT kinase is only activated on dimerization of the receptor, and KIT receptor heterodimers consisting of one normal KIT polypeptide and one abnormal KIT polypeptide are inactive Thus, patients heterozygous for these "dominant-negative" KIT missense mutations have only one-fourth of the normal amount of KIT receptor dimer, and accordingly they exhibit relatively severe piebald phenotypes In contrast, the second group of KIT mutations consists of proximally located nonsense, frameshift, and splice junction mutations that completely eliminate the production of KIT protein by the mutant gene Patients heterozygous for these "loss of function" mutant alleles thus express half of the normal amount of KIT receptor, resulting in haploinsufficiency for KIT -dependent signal transduction. These KIT mutations are thus associated with relatively mild piebald phenotypes, in which the depigmented patches are usually small and poliosis is often absent, although affected individuals frequently experience early graying of the hair In fact, some members of families with KIT mutations of this type have been so mildly affected that the clinical diagnosis was only made subsequent to DNA diagnosis Interestingly, we detected one of these proximal frameshifts, codons 250-251 DeltaCAGT, in three different, unrelated families with mild piebaldism (Spritz et al. , 1993), suggesting that this recurrent KIT gene mutation may account for a significant fraction of human piebaldism, especially among clinically milder cases The third group of mutations consist of frameshifts, nonsense, and splice junction mutations located distally, within the intracellular tyrosinase kinase domain These mutations tend to be associated with a rather variable phenotype, ranging from extremely mild to quite severe, even among affected members of an individual family All of these mutations would result in premature termination of translation, truncating the nascent KIT polypeptide distally, within the intracellular tyrosine kinase domain Clearly, these mutations would abolish expression of normal KIT polypeptide from this allele However, the truncated KIT receptors apparently can still bind SLF and even form dimers, dominant negatively inhibiting function of the normal KIT polypeptide (Lev et a!. , 1992) It is likely, however, that both the truncated KIT polypeptides and the incompletely translated KIr mRNA are relatively unstable Therefore, these mutations probably reduce KIT function to an amount between one-fourth and one-half of normal, accounting for the intermediate and highly variable piebald phenotype The fourth group of KIT mutations consists of only a single example, a missense substitution located within the extracellular ligand-binding domain of the KIT receptor. This substitution is not associated with any abnormality, and thus appears to constitute a normal polymorphic variant with no pathologic consequences (Ezoe et al, 1995). As noted above, this region of the KIT polypeptide consists principally of five immunoglobulin-like repeat domains, and it is therefore possible that this region may be at least in part functionally redundant Thus, many amino acid substitutions in this portion of the KIT polypeptide may have little or no effect on ligand binding, and thus may have little or no phenotypic effect. However, the extracellular portion of the KIT polypeptide is also thought to contain the segments that mediate receptor dimerization, and it seems likely that pathologic amino acid substitutions of residues involved in this process may be encountered in the future Overall, we have identified pathologic point mutations or large deletions of the KIT in approximately 75% of the patients with piebaldism studied to date (Ezoe et al., 1995). Of the remaining patients, none have abnormalities of the MGF gene (the human Steel gene homologue), suggesting that, in contrast with mice, mutations of this gene may not result in the piebald phenotype in humans (Ezoe et al , 1995) Genetic linkage analyses in two of these families showed complete linkage of the piebald trait to KIT intragenic markers (Ezoe et al, 1995), and some of these patients likely have occult KIT point mutations or gene deletions not detected by PCR-based SSCP/heteroduplex screening However, it is also possible that some might have abnormalities in the adjacent PDGFRA and KDR genes. The mouse "patch" (Ph) mutation, which results in a dominant white spotting phenotype similar to that of W, comprises a deletion that includes the pdgfra gene but not c-kit (Smith et al. , 1991 ; Stephenson et al. , 1991) Although white spotting in Ph mice might result either from deletion of the pdgfra gene itself or from inhibitory position effects of the large chromosomal deletion on expression of the nearby c-kit gene, it thus seems possible that some cases of human piebaldism might result from mutations in PDGFRA
Waardenburg syndrome, first described in 1951 by a Dutch ophthalmologist
(Waardenburg, 1951 ), is an autosomal dominant disorder characterized
clinically by piebald-like pigmentary anomalies of the skin and
hair, pigmentary abnormalities of the iris (heterochromia irides),
lateral displacement of the inner canthi of the eyes (dystopia canthorum),
and sensorineural deafness (Waardenburg, 1951; DiGeorge et al. ,
1960). Waardenburg syndrome occurs with an overall frequency of
1 to 2 per hundred thousand, and accounts for at least 0 5 percent
of cases of congenital deafness All of the abnormalities in Waardenburg
syndrome involve the neural crest, and both Hirschsprung disease
(Currie et al , 1986; Ariturk et al, 1992) and neural tube defects
(Bergleiter and Harris, 1992; Carezani-Gavin et al, 1992; Chatkupt
et al, 1993; Kromberg and Krause, 1993) occur at increased frequencies
among affected individuals Waardenburg syndrome has thus been considered
a more general disorder of neural crest development than piebaldism.
Three subtypes of Waardenburg syndrome have been distinguished on
clinical grounds; Waardenburg syndrome type I (WS 1) is the classic
form, Waardenburg syndrome type II (WS2; MIM #193510) lacks dystopia
canthorum, and Waardenburg syndrome type III (WS3; Klein-Waardenburg
syndrome; MIM #148820) is associated with limb abnormalities as
well as dystopia canthorum Elucidation of the molecular basis of
human WS 1 was facilitated by studies of a similar disorder of mouse,
"Splotch" (Sp) (reviewed in Pierpont and Erickson, 1993; Spritz,
1993). Splotch mutations typically result in piebald-like patches
of unpigmented skin and hair (Epstein et al, 1991, 1993; Goulding
et al, 1991; Moase and Trasler, 1992; Goulding et al, 1993; Vogan
et al, 1993), probably due to delayed migration of melanoblasts
or to a reduction in their number so that melanoblasts fail to reach
the affected regions before hair follicles develop The murine Splotch
locus encodes a developmental gene known as Pax-3, one of a family
of so-called "paired box" genes involved in embryological development
(Burri et al, 1989). Although the exact function of the Pax-3 gene
product is not yet known, it is thought to be a transcription factor
critical for activating melanoblasts to begin migration from the
neural crest The human P AX3 polypeptide contains four principal
structural motifs' the "paired-box" domain, a homeobox domain, a
conserved octapeptide, and a serine-threonineproline-rich carboxyl
segment (Figure 4) The 128-amino acid paired-box domain, from which
the name of the gene is derived, and the 60-amino acid homeobox
domain are both highly conserved amino acid sequence motifs present
in a number of mammalian and Drosophila genes involved in controlling
segmentation These motifs form a helix-turn-helix structure and
are both thought to mediate DNA binding 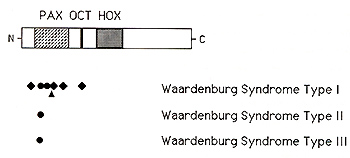
The murine sptolch locus was mapped to chromosome 1, in a region that is homologous to part of human chromosome 2q, and the mouse Sp2H mutation was then found to consist of a deletion in the paired box domain of Pax-3 (Epstein el at. , 1991 ). The human WS 1 gene was first localized to the distal long arm of chromosome 2 on the basis of a patient with an chromosomal inversion of 2q35-q373 (Ishikiriyama el at., 1989), and subsequent genetic linkage analyses demonstrated linkage between WS I and markers in this region (Foy el at. , 1990; Asher el at. , 1991 ), suggesting that human WS I and mouse Splotch might be homologous Although only a portion of the human P AX3 gene had been characterized, Tassabehji and coworkers ( 1992) and Baldwin and coworkers ( 1992) quickly identified point mutations in several patients with WS I, and demonstrated that these mutations were genetically linked to the WS I phenotype in these families At least seven different mutations of the PAX3 gene have now been reported in patients with WS I, including an in-frame deletion within the paired-box domain (Tassabehji el al., 1992), three frameshifts (Morell el al., 1992, 1993; Tassabehji el al., 1993), and two missense substitutions, both located within the paired box domain (Baldwin el al. , 1992; Hoth el al., 1993) Furthermore, Hoth el al. (1993) identified an additional missense mutation, N47H, in a family with so-called WS3, demonstrating that WS I and WS3 are allelic, resulting from different abnormalities of PAX3 By genetic linkage analysis of 41 families, Farrer and coworkers ( 1992) determined that abnormalities on distal chromosome 2, presumably in the p AX3 gene, account for about half of cases of WS 1. This implies the existence of at least one additional gene for Waardenburg syndrome located elsewhere in the genome. In addition, they found no evidence of linkage between chromosome 2 markers and WS2, indicating that this disorder does not result from mutations of PAX3 However, Tassabehji et al. (1993) reported a P AX3 missense substitution in a family that they considered to have WS2, although clinical photographs of this family are suggestive of mild dystopia canthorum and thus WS 1 Two groups reported refined map data for the human PAX3 gene Tsukamoto and coworkers ( 1992) showed that the distal chromosome 2q inversion associated with Waardenburg syndrome type I, described above, interrupted exons 2 and 3 of the P AX3 gene, and they suggested that the PAX3 gene is located within chromosome segment 2q35 Similarly, Lu-Kuo et al. (1993) showed that the PAX3 gene is deleted in a patient with Waardenburg syndrome and a distal chromosome 2 deletion (Kirkpatrick et al., 1992), and they mapped the PAX3 gene to 2q361-q362. These two map assignments are actually quite close, probably within the limits of cytogenetic resolution Farrer and coworkers ( 1992) found no evidence of genetic linkage between chromosome 2 markers and WS2, indicating that WS2 does not usually result from mutations of P AX3 Recently, Hughes and coworkers ( 1994) reported localization of the gene for WS2 to chromosome segment 3p 12-p 14.1, in close proximity to MJTF, the human homologue to the mouse microphthalmia gene (Tachibana et al, 1994). The phenotype ofmi mutant mice includes both pigmentary abnormalities and hearing loss, as well as microphthalmia, various abnormalities of neural crest development, deficient mast cells, and osteopetrosis (Silvers, 1979; Lyon and Searle, 1989), suggesting that MITF is a very good candidate for the human WS2 gene. The MITF gene product is a novel member of the basic-helix-loop-helix-leucine zipper of transcription factors (Hodgkinson et al., 1993; Tachibana et al, 1994)--proteins that have a variety of roles in regulating gene expression, cellular proliferation and differentiation-consistent with the pleiotropic phenotypic effects seen in mi mutant mice and humans with WS2 Thus, it seems likely that at least some cases of human WS2 result from mutations of MITF. In a surprising turn of events, Barr and coworkers ( 1993) showed that the P AX3 gene is rearranged in the chromosome 2; 13 translocation consistently observed in cases of alveolar rhabdomyosarcoma (MIM #268220), an aggressive solid tumor of childhood. In three such tumors the chromosome 2 breakpoints are located within the fourth intervening sequence of the PAX3 gene, juxtaposing the upstream PAX3 DNA binding motifs, including the paired box domain, conserved octapeptide, and homeobox domain, to a specific gene located on chromosome 13q 14 As the result, an abundant novel mRNA is transcribed, most likely encoding a novel fusion protein that interferes with the normal controls of striated muscle cell proliferation Thus, the PAX3 gene appears to lead a double life. inherited loss-of function mutations result in Waardenburg syndrome and acquired gain of function mutations result in alveolar rhabdomyosarcoma.
This work was supported by a grant from the National Institutes of Health (AR-39892) and By a Clinical Research Grant from the March of Dimes Birth Defects Foundation (60281 ). This is paper number 3426 from the Laboratory of Genetics, University of Wisconsin.
Andria G, Ballabio A, Parenti G (1987) X-Iinked ichthyosis due to steroid sulfatase deficiency associated with hypogonadism and anosmia Ann Neuro. 22.98, Ariturk E, Tosyali N, Ariturk N (1992) A case ofWaardenburg syndrome and aganglionosis Turk J Pediatr 34'111-114 Asher JH Jr, Morell R, Friedman TB (1991) Waardenburg syndrome (WS)' The analysis ofa single family with a WS 1 mutation showing linkage to RFLP markers on human chromosome 2q. Am J Hum Genet 48'43-52 Baldwin CT, Hoth CF, Amos JA, Da Silva EO, Milunsky A (1992) An exonic mutation in the HuP2 paired domain gene causes Waardenburg's syndrome. Nature 355'637-638, Barr FG, Galili N, Holick J, Biegelo JA, Rovera G, Emanuel BS (1993) Rearrangement of the P AX3 paired box gene in the paediatric solid tumor alveolar rhabdomyosarcoma Nature Genetics 3:113-117 Bennett DC, Huszar D, Laipis PJ, Jaenisch R, Jackson IJ (1990) Phenotypic rescue of mutant brown melanocytes by a retrovirus carrying a wild-type tyrosinase-related protein gene Development 110 471-475 Bergen AAB, Samanns C, Van Dorp DB, Ferguson-Smith MA, Gal A, Bleeker-Wagemakers EM (1990), Localization of the X-Iinked ocular albinism gene (OA1) between DXS278/DXS237 and DXS 143/DXS 16 by linkage analysis Ophthalmic Paediat Genet 11'165-170, Bergen AAB, Samanns C, Schuurman ElM, Van Osch L, Van Dorp DB, Pinckers AJLG, Bakker E, Gal A, Van Ommen GJB, Bleeker-Wagemakers EM (1991) Multipoint linkage analysis in X-Iinked ocular albinism of the Nettleship-Falls type, Hum Genet 88'162-166, Bergleiter ML, Harris Dl (1992) Waardenburg syndrome and meningocele Am J Med Genet 44'541 Besmer P, Murphy JE, George PC, Qiu F, Bergold PJ, Lederman L, Snyder HW Jr, Brodeur D, Zuckerman EE, Hardy WS (1986) A new acute transforming feline retrovirus and the relationship of its oncogene v-kit with the protein kinase gene family Nature 320:415-421, Boissy RE, Zhao H, Austin LM, Nordlund 11, King RA (1993) Melanocytes from an individual with brown oculocutaneous albinism lack expression of TRP-1 , the product of the human homologue of the murine brown locus Am J Hum Genet 53' Suppl, 160 Breathnach AS. Fitzpatrick TB, Wyllie LMA (1965) Electron microscopy ofmelanocytes in a case of human piebaldism J Invest Dermatol 4528-37 Burri M, Tromvoukis Y, Bopp D, Frigerio G, No11 M (1989) Conservation of the paired domain in metazoans and its structure in three isolated human genes EMBO J 81183-1190 Butler MG (1989) Hypopigmentation a common feature of Prader-Labhart-Willi syndrome Am J Hum Genet 45140-146, Carezani-Gavin M, Slarren SK, Steege T (1992) Waardenburg syndrome associated with meningomyelocele Am J Med Genet 42135 Chabot B, Stephenson DA, Chapman VM, Besmer P, Bernstein A ( 1988) The proto oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse Wlocus Nature 335'88-90 Chakraborty AK, Orlow SI, Pawelek IM ( 1992) Evidence that DOP Achrome tautomerase is a ferrous iron-binding glycoprotein FEB S Lett 302126-128 Charles SI, Moore AT, Yates JRW (1992) Genetic mapping of X linked ocular albinism linkage analysis in British families I Med Genet 29552-554 Chatkupt S, Chatkupt S, Johnson WG (1993) Waardenburg syndrome and myelomeningocele in a family I Med Genet 3083-84 Chintamaneni CD, Ram say M, Colman MA, Fox MF, Pickard RT, Kwon BS (1991) Mapping the human CAS2 gene, the homologue of the mouse brown (b) locus, to human chromosome 9p22-pter Biochem Biophys Res Commun 178 227-235. Cohen T, Muller RM, Tomita Y, Shibahara S Nucleotide sequence of the cDNA encoding human tyrosinase-related protein Nucl Acids Res 182807 -2808 ( 1990) Cooke IV (1952) Familial white skin spotting (piebaldness) ('partial albinism') with white forelock I Pediatr 41 -12. Copeland NG, Gilbert Dl, Cho BC, Donovan PI, Jenkins NA, Cosman D, Anderson D, Lyman SD, Williams DE (1990) Mast cell growth factor maps near the steel locus on mouse chromosome 10 and is deleted in a number of steel alleles. Cell 63.175-183 Currie ABM, Haddad M, Honeyman M, Boddy S-A M (1986) Associated developmental abnormalities of the anterior end of the neural crest Hirschsprung's disease Waardenburg's syndrome I Pediatr Surg 21 248-250 d' Auriol L, Mattei MG, Andre C, Galibert F ( 1988) Localization of the human c-kit protooncogene on the q11-q12 region of chromosome 4 Hum Genet 78:374-376 DiGeorge AM, Olmsted RW, Harley RD (1960) Waardenburg syndrome I Pediatrics 57.649 669 Durham FM (1904) On the presence of tyrosinases in the skins of some pigmented vertebrates Proc R Soc Lond 74.310-313 Epstein Dl, Vekemans M, Gros P (1991) Splotch (Sp2H), a mutation affecting development of the mouse neural tube, shows a deletion within the paired homeodomain of Pax-3 Ce1l67767-774 Epstein Dl, Vogan KI, Trasler DG, Gros P (1993) A mutation within intron 3 of the Pax-3 gene produces aberrantly spliced mRNA transcripts in the splotch (Sp) mouse mutant Proc Natl Acad Sci USA 90;532-536 Ezoe K, Holmes SA, Ho L, Bennett CP, Bolognia IL, Brueton L, Burn I, Falabella R, Gat to EM, Ishii N, Moss C, Pittelkow MR, Thompson E, Ward KA, Spritz RA (1995) Novel mutations and deletions of the KIT (steel factor receptor) gene in human piebaldism. Am I Hum Genet, in press Falik-Borenstein TC, Holmes SA, Borochowitz Z, Spritz RA (1995) DNA-based carrier detection and prenatal diagnosis of tyrosinase-negative oculocutaneous albinism (OCA 1 A) Prenatal Diagnosis, in press Farrer LA, Grundfast KM, Amos I, Arnos KS, Asher IH Ir, Beighton P, Diehl SR, Fex J, Foy C, Friedman TB, Greenberg I, Hoth C, Marazita M, Milunsky A, Morell R, Nance W, Newton V, Ramesar R, San Agustin TB, Skare I, Steens CA, Wagner RG Ir, Wilcox ER, Winship I, Read AP (1992) Waardenburg syndrome (WS) type I is caused by defects at multiple loci, one of which is near ALPP on chromosome 2 First report of the WS consortium Am I Hum Genet 50:902-913 Flanagan IG, Leder P ( 1990) The kit ligand a cell surface molecule altered in steel mutant fibroblasts Cell 63:185-194. Fleischman RA, Saltman DL, Stastny V, Zneimer S ( 1991) Deletion of the c-kit protooncogene in the human developmental defect piebald trait Proc Natl Acad Sci USA 8810885-10889 Fleischman RA ( 1992) Human piebald trait resulting from a dominant negative mutant allele of the c-kit membrane receptor gene 1 Clin Invest 89:1713-1717 Foy C, Newton VE, Wellesley D, Harris R, Read AP (1990) Assignment of WS 1 locus to human 2q37 and possible homology between Waardenburg syndrome and the Splotch mouse Am 1 Hum Genet 46.1017-1023 Froggat P (1951) An outline with bibliography of human piebaldism Ir 1 Med Sci 39886-94 Fukai K, Holmes SA, Lucchese N1, Siu VM, Weleber RG, Schnur RE, Spritz RA (1995) Autosomal recessive ocular albinism associated with a functional significant tyrosinase gene polymorphism Nature Genet 9:92-95 Funderburk S1, Crandall BF (1974) Dominant piebald trait in a retarded child with a reciprocal translocation and small intercalary deletion Am 1 Hum Genet 26 715-722. Gardner JM, Nakatsu Y, Gondo Y, Lee S, Lyon MF, King RA, Brilliant MH (1992) The mouse pink-eyed dilution gene association with human Prader-Willi and Angelman syndromes. Science 257.1121-1124. Garrod AE ( 1908) The Croonian lectures on inborn errors of metabolism lecture I. Lancet 21-7 Geissler EN, Ryan MA, Housman DE (1988) The dominant-white spotting (W) locus of the mouse encodes the c-kit proto-oncogene Cell 55185-192 Gellius ( 1952) The Attic Nights of Aulus Gellius Book 9 Rolfe 1C, trans Cambridge, Harvard University Press Giebel LB, Spritz RA ( 1990) RFLP for MboI in the human tyrosinase (TYR) gene detected by PCR Nucl Acids Res 183103. Giebel LB, Spritz RA (1991) Mutation of the c-kit (mast/stem cell growth factor receptor) proto-oncogene in human piebaldism Proc Natl Acad Sci USA 88'8696-8699 Giebel LB, Strunk KM, King RA, Hanifin 1M, Spritz RA (1990) A frequent tyrosinase gene mutation in classic, tyrosinase-negative (type IA) oculocutaneous albinism Proc Natl Acad Sci USA 873255-3258 Giebel LB, Strunk KM, Spritz RA ( 1991a) Organization and nucleotide sequences of the human tyrosinase gene and a truncated tyrosinase-related segment. Genomics 9435 445 Giebel LB, Tripathi RK, King RA, Spritz RA ( 1991 h) A tyrosinase gene missense mutation in temperature-sensitive type I oculocutaneous albinism A human homologue to the Siamese cat and the Himalayan mouse 1 Clin Invest 87.1119-1122. Giebel LB, Strunk KM, Holmes SA, Spritz RA (1992) Organization and nucleotide sequence of the human KIT (mast/stem cell growth factor receptor) proto-oncogene. Oncogene 72207-2217. Goulding MD, Chalepakis G, Deutsch U, Erselius 1R, Gruss P (1991) Pax-3, a novel murine DNA binding protein expressed during early neurogenesis EMBO 1 10 1135-1147. Goulding MD, Sterrer S, Fleming 1, Balling R (1993) Analysis of the Pax-3 gene in the mouse mutant splotch Genomics 17:355-363 Hodgkinson CA, Moore K1, Nakayama A, Steingrimsson E, Copeland NG, NA, Arnheiter H ( 1993) Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein Cell 74:395-404 Hoo 11, Haslam RHA, van Orman C (1986) Tentative assignment of piebald trait gene to chromosome band 4q 12 Hum Genet 73 230-231 Hoth CF, Milunsky A, Lipsky N, Sheffer R, Clarren SK, Baldwin CT (1993) Mutations in the paired domain of the human PAX3 gene cause Klein-Waardenburg syndrome (WS-III) as well as Waardenburg syndrome type I (WS-I) Am J Hum Genet 52-455-462 Hu F Hanifin JM, Prescott GH, Tongue AC (1980) Yellow mutant albinism cytochemical, ultrastructural, and genetic characterization suggesting multiple allelism Am J Hum Genet 32-387-395 Huang E, Nocka K, Beler DR, Chu TY, Buck J, Lahm HW, Wellner D, Leder P, Besmer P ( 1990) The hematopoietic growth factor KL is encoded by the SI locus and is the ligand of the c-kit receptor, the gene product of the Wlocus Cell 63-225-233 Hughes AE, Newton YE, Liu XZ, Read AP (1994) A gene for Waardenburg syndrome type 2 maps close to the human homologue of the microphthalmia gene at chromosome 3p12-p14-1 Nature Genetics 7509-512 Ishikiriyama S, Tonoki H, Shibuya Y, Chin S, Harada N, Abe K, Niikawa N (1989) Waardenburg syndrome type I in a child with de novo inversion (2)(q35q373) Am J Med Genet 33 505-507 Jackson IJ ( 1988) A cDNA encoding tyrosinase-related protein maps to the brown locus in mice Proc Natl Acad Sci USA 85-4392-4396 limbow K, Fitzpatrick TB, Szabo G, Hori Y (1975) Congenital circumscribed hypomelanosis a characterization based on electron microscopic study of tuberous sclerosis, nevus depigmentosus, and piebaldism l Invest Dermatol 64:50-62 liminez-Cervantes C, Solano F, Kobayashi T, Hearing Vl, Lozano JA, Garcia-Borron lC (1994) The DHICA oxidase activity of mouse melanoma tyrosinases l BioI Chem, in press Keeler CE ( 1934) The heredity of a congenital white spotting in Negroes lAm Med Assoc 103 179-180 King RA, Mentink MM, Getting WS ( 1991) Nonrandom distribution of missense mutations within the human tyrosinase gene in type I (tyrosinase-related) oculocutaneous albinism Mol BioI Med 819-29 Kirkpatrick Sl, Kent CM, Laxova R, Sekhon GS (1992) Waardenburg syndrome type I in a child with deletion (2)(q35q362) Am l Med Genet 44 699- 700 Kromberg JGR, Krause A (1993) Waardenburg syndrome and spina bifida Am J Med Genet 45-536-537 Kugelman TP, van Scott El ( 1961) Tyrosinase activity in melanocytes of human albinos J Invest Dermato137 73-76 Kwon BS, Haq AK, Pomerantz SH, Halaban R (1987) Isolation and sequence of a putative cDNA clone for human tyrosinase that maps at the mouse c-albino locus Proc Natl Acad Sci USA 84-7473-7477 Lacassie Y, Thurmon TF, Tracy MD, Pelias MZ (1977) Piebald trait in a retarded child with interstitial deletion of chromosome 4 Am l Hum Genet 29641-642 Lee S- T , Nicholls RD, Schnur RE, Guida LC, Lu-Kuo l, Spinner NB, Zackai EH, Spritz RA (1994) Diverse mutations of the p gene among African-Americans with type II (tyrosinase-positive) oculocutaneous albinism (GCA2) Hum Molec Genet 3 2047 2051 Lee S- T , Strunk KM, Bundey S, Laxova R, Musarella M, Spritz RA ( 1994) Mutations of the p gene in oculocutaneous albinism, ocular albinism, and Prader-Willi syndrome plus albinism New Engl l Med 330529-534 Lerch K ( 1988) Protein and active-site structure of tyrosinase Adv Pigment Cell Res, Prog Clin BioI Res 25685-98 Lee, S-T, Nicholls RD, Jong MTC, Spritz RA (1995) Organization and sequence of the human P gene and identification of a new family of transport proteins, Genomics, in press, Lerch K ( 1988) Protein and active-site structure of tyrosinase Adv Pigment Cell Res, Prog Clin BioI Res 25685-98 Lev S, Yarden Y, Givol D ( 1992) Dimerization and activation of the kit receptor by monovalent and bivalent binding of the stem cell factor, J BioI Chem 267'15970-15977 Lucian (1905) The Works of Lucian of Samosata, vol, 1 Fowler HW, trans], Oxford, Clarendon Press, Lu-Kuo J, Ward DC, Spritz RA (1993) Fluorescence in situ hybridization mapping of 25 markers on distal human chromosome 2q surrounding the human Waardenburg syndrome, type I (WS1) locus (PAX3 gene) Genomics 16 173-179, Lyon M, Searle AG ( 1989) Genetic Variants and Strains of the Laboratory Mouse, Oxford University Press, Oxford Martinez JH, Solano F, Garcia-Borron JC, Iborra JL, Lozano JA (1985) The involvement of histidine at the active site of Harding-Passey mouse melanoma tyrosinase, Biochem Int 11'729- 738, Moase CE, Trasler DG ( 1992) Splotch locus mouse mutants' models for neural tube defects and Waardenburg syndrome type I in humans, J Med Genet 29'145-151, Morell R, Friedman TB, Moelijopawiro S, Hartono, Soewito, Asher JH Jr (1992) A frameshift mutation in the HUP2 paired domain of the probable human homolog of murine PAX-3 is responsible for Waardenburg syndrome type I in an Indonesian family Hum Mol Genet 1 243-247 Morell R, Friedman TB, Asher JH Jr ( 1993) A plus-one frameshift mutation in P AX3 alters the entire deduced amino acid sequence of the paired box in a Waardenburg syndrome type 1 (WS1) family, Hum Molec Genet 21487-1488 Morgan J (1786) Some accounts of motley colored or pye Negro girl and mulatto boy, Trans Am Philos Soc (Phila) 2392-395, Morrison-Graham K, Takahashi Y (1993) Steel factor and c-kit receptor' from mutants to a growth factor system BioEssays 15'77-83 Murphy M, Reid K, Williams DE, Lyman SD, Bartlett PF (1992) Steel factor is required for maintenance, but not differentiation, of melanocyte precursors in the neural crest Devel BioI 153 396-401 Murty VVVS, Bouchard B, Mathew S, Vijayasaradhi S, Houghton AN (1992) Assignment of the human TYRP (br'own) locus to chromosome region 9p23 by nonradioactive in Situ hybridization Genomics 13 227-229 Nance WE, Jackson CE, Witkop CJ Jr (1970) Amish albinism' a distinctive autosomal recessive phenotype Am J Hum Genet 22579-586, Nicholls RD (1993) Genomic imprinting and uniparental disomy in Angelman and Prader-Willi syndromes' A review, Am J Med Genet 4616-25 Nishikawa S, Kusakabe M, Yoshinaga K, Ogawa M, Hayashi S-I, Kunisada T, Era T, Sakakura T, Nishikawa S- I ( 1991) Rn utero manipulation of coat color formation by a monoclonal anti-c-kit antibody two distinct waves of c-kit-dependency during melanocyte development EMBO J 10 2111-2118 Online Mendelian Inheritance in Man, OMIM (TM) [database online] (1994) The Johns Hopkins University, Baltimore, Maryland Pierpont JW, Erickson RP (1993) Facts on PAX Am J Hum Genet 52451-454 Pliny ( 1942) The Natural History of Pliny. Plinius Secundus the Elder. Book 7. Rackman H, trans Cambridge, Harvard University Press Ramsay M, Colman M-A, Stevens G, Zwane E, Kromberg J, Farrall M, Jenkins T (1992) The tyrosinase-positive oculocutaneous albinism locus maps to chromosome 15q 11.2-q 12 Am J Hum Genet 51 879-884 Reith AD, Rottapel R, Giddens E, Brady C, Forrester L, Bernstein A (1990) W mutant mice with mild or severe developmental defects contain distinct point mutations in the kinase domain of the c-kit receptor Genes & Devel 4 390-400 Rinchik EM, Bultman SJ, Horsthemke B, Lee S-T, Strunk KM, Spritz RA, Avidano KM, Jong MTC, Nicholls RD ( 1993) A gene for the mouse pink-eyed dilution locus and for human type II oculocutaneous albinism. Nature 361 72- 76 Schaefer L, Ferrero GB, Grillo A, Bassi MT, Roth EJ, Wapenaar MC, Van Ommen GJB, Mohandas TK, Rocchi M, Zoghbi HY, Ballabio A ( 1993) A high resolution deletion map of human chromosome Xp22 Nature Genetics 4272-279. Schnur RE, Nussbaum RL, Anson-Cartwright L, McCowell C, Worton RG, Musarella MA ( 1991 ) Linkage analysis in X-Iinked ocular albinism Genomics 9605-613 Shibahara S, Tomita Y, Sakakura T, Nager C, Chaudhuri B, Muller R (1986) Cloning and expression ofcDNA encoding mouse tyrosinase Nucl Acids Res 142413-2427 Silvers WK (1979) The Coat Colors ofMice Springer-Verlag, New York Smith EA, Seldin MF, Martinez L, Watson ML, Choudhury GG, Lalley PA, Pierce J, Aaronson S, Barker J, Naylor SL, Sakaguchi AY (1991) Mouse platelet-derived growth factor receptor alfa gene is deleted in W19H and patch mutations on chromosome 5 Proc Natl Acad Sci USA 884811-4815 Spritz, RA ( 1993 ) Molecular basis of piebaldism and Waardenburg's syndrome Current Opinion in Dermatology 1 78-93 Spritz RA ( 1994a) Molecular genetics of oculocutaneous albinism Hum Molec Genet 3.1469-1475 Spritz RA ( 1 994b) The molecular basis of human piebaldism J Invest Dermatol, in press Spritz RA and Hearing VJ Jr (1995) Genetic disorders of pigmentation Ann Rev Genet 221 45 Spritz RA, Strunk KM, Giebel LB, King RA ( 1990) Detection of mutations in the tyrosinase gene in a patient with type IA oculocutaneous albinism New Engl J Med 322 1724 1728 Spritz RA, Droetto S, Fukushima Y (1992) Deletion of the KIT and PDGFRA genes in a patient with piebaldism. Amer J Med Genet 44.492-495 Spritz RA, Holmes SA, Berg SZ, Nordlund JJ, Fukai K (1993) A recurrent deletion in the KIT (mast/stem cell growth factor receptor) gene is a frequent cause of human piebaldism Hum Molec Genet 2.1499- 1500 Spritz RA, Ho L, and Strunk KM ( 1 994a) Inhibition of proliferation of human melanocytes by a KIT antisense oligodeoxynucleotide Implications for human piebaldism and mouse dominant white spotting ( W). J Investig Derm 103.148- 150 Spritz, RA, Lu-Kuo, JM, Le Paslier, DL, Altherr, MR, Dorman, T, Moir, DT, Strunk, KM, and Ward, DC (1 994b) A 2-Mb Y AC contig spanning a cluster of genes encoding type III receptor tyrosine kinases (PDC;FRA-KIT-KDR) is located at 4q12 Genomics 22431-436 Steel KP, Davidson DR, Jackson IJ (1992) TRP2/DT, a new early melanoblast marker, shows that steel growth factor ( c-kit ligand) is a survival factor Development 1 15 1111-1119 Stephenson DA, Mercola M, Anderson E, Wang C, Stiles CD, Bowen-Pope DF, Chapman VM (1991) Platelet-derived growth factor receptor alfa-subunit gene (Pdgfra) is deleted in the mouse patch (Ph) mutation Proc Natl Acad Sci USA 886-10. Summers CG, Creel D, Townsend D, King RA (1991) Variable expression of vision in sibs with albinism Am 1 Med Genet 40327-331. Sunohara N, Sakuragawa N, Satoyoshi E, Tanae A, Shapiro Ll (1986) A new syndrome of anosmia, ichthyosis, hypogonadism, and various neurological manifestations with deficiency of steroid sulfatase and aryl sulfatase C Ann Neurol 19174-181 Tachibana M, Perez-lurado LA, Nakayama A, Hodgkinson CA, Li X, Schneider M, Miki T , Fex l, Francke U, Arnheiter H (1994) Cloning ofMlTF., the human homolog of the mouse microphthalmia gene, and assignment to human chromosome 3, region p141p123 Hum Molec Genet, in press. Tan lC, Nocka K, Ray P, Traktman P, Besmer P (1990) The dominant W42 spotting phenotype results from a missense mutation in the c-kit receptor kinase Science 247.209-212 Tassabehji M, Read AP , Newton VE, Harris R, Balling R, Gruss P, Strachan T (1992) Waardenburg's syndrome patients have mutations in the human homologue of the Pax3 paired box gene Nature 355.635-636 Tassabehji M, Read AP, Newton YE, Pat ton M, Gruss P, Harris R, Strachan T (1993) Mutations in the PAX3 gene causing Waardenburg syndrome type 1 and type 2 Nature Genetics 3 26-30 Tomita Y, Takeda A, Okinaga S, Tagami H, Shibahara S (1989) Human oculocutaneous albinism caused by single base insertion in the tyrosinase gene. Biochem Biophys Res Commun 164990-996. Tripathi RK, Giebel LB, Strunk KM, Spritz RA (1991) A polymorphism of the human tyrosinase gene that is associated with temperature-sensitive enzymatic activity. Gene Expression 1.103-110. Tripathi RK, Strunk KM, Giebel LB, Weleber RG, Spritz RA (1992) Tyrosinase gene mutations in type I (tyrosinase-deficient) oculocutaneous albinism define two clusters of missense substitutions Amer 1 Med Gen 43 865-871. Tsukamoto K, Tohma T, Ohta T, Yamakawa K, Fukushima Y, Nakamura Y, Niikawa N ( 1992) Cloning and characterization of the inversion breakpoint at chromosome 2q35 in a patient with Waardenburg syndrome type I Hum Molec Genet 1 315-317. Vandenbark GR, DeCastro CM, Taylor H, Dew-Knight S, Kaufman RE (1992) Cloning and structural analysis of the human c-kit gene Oncogene 7.1259-1266 Vijayasaradhi S, Bouchard B, Houghton AN (1990) The melanoma antigen gp75 is the human homologue of the mouse b (brown) locus gene product 1 Exp Med 171.1375-1380 Vogan Kl, Epstein Dl, Trasler DG, Gros P (1993) The splotch-delayed (Spd) mouse mutant carries a point mutation within the paired box of the Pax-3 gene Genomics 17 364- 369 Waardenburg Pl (1951) A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair with congenital deafness Am 1 Hum Genet 3195-253 Wagstaff 1 ( 1993) A translocation-associated deletion defines a critical region for the 9p syndrome Am 1 Hum Genet 53, Suppl, 619. Wapenaar MC, Bassi MT, Schaefer L, Grillo A, Ferrero GB, Chinault AC, Ballabio A, Zoghbi HY (1993) The genes for X-Iinked ocular albinism (OA1) and microphthalmia with linear skin defects (MLS) cloning and characterization of the critical regions Hum Molec Genet 2947-957 Wildenberg SC, Boissy RE, Getting WS, Fryer JP, Zhao H, King RA (1994) Identification of a mutation in the tyrosinase related protein 1 (TRP 1) gene associated with brown oculocutaneous albinism (GCA3) Am J Hum Genet 55.Suppl. A5 Winder AJ, Kobayashi T, Tsukamoto K, Urabe K, Aroca P, Kameyama K, Hearing VJ (1994) The tyrosinase gene family. Interactions of melanogenic proteins to regulate melanogenesis. Biochem J, in press. Winship IM, Babaya M, Ramesar RS (1993) X-Iinked ocular albinism and sensorineural deafness linkage to Xp22.3. Genomics 18444-445. Witkop CJ Jr, Quevedo WC Jr, Fitzpatrick TB, King RA (1989) Albinism. In, Scriver CR, Beaudet AL, Sly WS, Valle D, eds The Metabolic Basis of Inherited Disease New York, McGraw-Hill, pp 2905-2947 Wittbjer A, Dahlback B, Gdg G, Rosengren A-M, Rosengren E, Rorsman H ( 1989) Isolation of human tyrosinase from cultured melanoma cells. Acta Derm Venereol (Stokh) 69.125-131. Yamamoto Y, Nishimoto H, Ikemoto S ( 1989) Interstitial deletion of the proximal long arm of chromosome 4 associated with father-child incompatibility within the Gc-system probable reduced gene dosage effect and partial piebald trait. Am J Med Genet 32.520-523. Yarden Y, Kuang W-J, Yang-Feng T, Coussens L, Munemitsu S, Dull TJ, Chen E, Schlessinger J, Francke U, Ullrich A ( 1987) Human proto-oncogene c-k;t. a new cell surface receptor tyrosine kinase for an unidentified ligand EMBG J 63341-3351. Zsebo KM, Williams DA, Geissler EN, Broudy VC, Martin FH, Atkins HL, Hsu RY, Birkett NC, Gkino KH, Murdock DC, Jacobsen FW, Langley KE, Smith KA, Takeishi T, Cattanach BM, Galli SJ, Suggs SV ( 1990) Stem cell factor is encoded at the SIlocus of the mouse and is the ligand for the c-k;t tyrosine kinase receptor. Cell 63.213-224 |