|
Centre for Genome Research, University of Edinburgh,
Kings Buildings, West Mains Rd. Edinburgh.
Gene Trapping in Embryonic Stem Cells. Embryonic stem (ES) cell
lines isolated from the inner cell mass of the mouse blastocyst
(Evans & Kaufman, 1983), can be maintained as pluripotent stem cells
and genetically manipulated in vitro. When reintroduced into a host
embryo they can contribute to all tissues, including the germline
of the resultant chimera (Beddington & Robertson, 1989). ES cells
have provided the essential tool in the genetic manipulation of
the mouse genome whereby an alteration to an allele of a loci can
be made in vitro, introduced into the germline and then bred to
homozygosity where the phenotypic effect can be examined. Gene trapping
in ES cells provides a method to identify and functionally characterise
novel genes and has proven very powerful in the molecular analysis
of embryonic development (Gossler et al., 1989). The design and
uses of the different types of gene trap constructs has been reviewed
(Skarnes, 1993; Hill & Wurst, 1993). Briefly, the important features
of a gene trap vector is an easily assayable reporter gene ( eg.
the gene encoding the bacterial enzyme, B-galactosidase (lacZ)))
linked to an upstream splice acceptor site and a selectable marker
(Figure 1). When the construct integrates into the intron of an
active gene a fusion transcript is generated containing exons of
the trapped gene that lie upstream of the integration site and the
lacZ reporter sequence (Skarnes, Auerbach & loyner, 1992). There
are several consequences of this event. First, the reporter gene
expression is under the control of the regulatory elements of the
trapped endogenous gene and the simple analysis of LacZ expression
can provide clues to the function of the trapped gene. Second, the
endogenous gene can be cloned from the fusion transcript using RACE-PCR
(rapid amplification of cDNA endspolymerase chain reaction) and
primers complementary to the known lacZ sequences. Sequence information
can provide further clues as to the gene's cellular function particularly
if the gene shows homology to known gene families. Finally, the
insertional event has the potential to be mutagenic and so the introduction
of the integration into the mouse germline and the subsequent breeding
to homozygosity allows the functional role of the gene to be addressed
in vivo . Trapping vectors must also employ a selectable marker,
most commonly the gene encoding neomycin phosphotransferase (neoR),
which can either be expressed from a constitutive promoter ( eg.
the promoter for the glycolytic enzyme phosphoglycerate kinase (PGK-l)),
or coexpressed as a fusion product (�geo) with LacZ (Freidrich &
Soriano, 1991). In the latter case, the expression of the selectable
marker is also under the control of the endogenous trapped gene's
control elements. The advantage of using �geo is that cell clones
containing true gene trap events can be selected directly in the
drug, G418. However, only integrations into genes that are expressed
in undifferentiated ES cells will be selected. When an independently
driven promoter is used for the expression of the selectable marker,
all random integrations must be 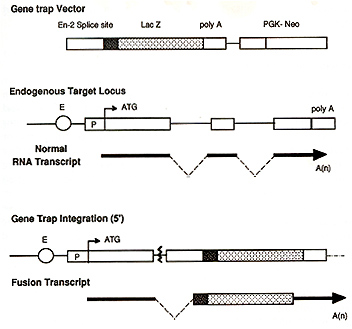
screened to identify true gene trap events, the frequency of which can be as lowas 2-5%. However, the advantage of this strategy is that it is possible to identify and fully characterise gene trap integrations into a class of genes that are expressed in differentiated, but not undifferentiated ES cells. This strategy was used in the study described here.
In a random gene trapping approach the ES cell clones were selected on the basis of reporter gene expression in undifferentiated ES cells and then screened for expression in the developing embryos. Integrations that result in the expression of the reporter gene in the tissue or developmental pathway of interest can then be selected for further study. In a large screen of over 250 gene trap integrations, 30% were constitutively expressed in the developing embryo, 30% were expressed in a developmentally-regulated manner and 30% were not expressed at the stages analysed (Wurst et al, 1994) .This is a particularly time consuming and expensive in viva screening strategy and clearly an in vitra pre-screen that could select for integrations into genes expressed in cell lineages of interest would be very useful.
ES cells can differentiate into a large number of different lineages in vitra (Doetschman etal. 1985). Therefore it should be possible to screen for gene trap integrations into an endogenous trapped gene that is expressed in a cell lineage of interest. We have performed a directed gene trap screen where gene trap integrations were selected in vitra on the basis of their response to the addition of the morphogen retinoic acid (RA). Addition of exogenous RA is known to have a profound effect on embryogenesis (reviewed by Eichele, 1989) and is known to activate, in vitra (Simeone etal. 1990,1991) and in viva, (Conlon & Rossant, 1992) well-characterised developmentally-regulated genes. Thus one would predict that genes that respond to RA would be expressed in a developmentally regulated manner .
Gene Trap Vector. The gene trap vector used was based on that described by Gossler et al, 1989. Briefly, PT1-ATG contained the En-2 splice acceptor site fused to the lacZ reporter gene which included an ATG start site. The bacterial neomycin resistance gene (neoR), driven by the phosphoglycerate kinase (PGK-1) promoter, was used as a selectable marker.
R1 ES cells (Nagy et al. 1993) were maintained on primary embryonic fibroblasts as described (Wurst & ]oyner, 1993). After electroporation and selection in G418 for 8 days, resistant colonies were replica-plated (Hill & Wurst, 1993) and the filters were placed in ES cell medium (without LIP) containing 5% FCS and 10 high �6 M all-trans retinoic acid for 48 hours. Medium was replaced with fresh RA-containing medium 6 hours prior to staining. After staining for B-galactosidase activity, blue colonies were picked from the master plate, ES cell clones were expanded and retested for B-gal activity in the presence and absence of RA and clones were examined microscopically. Clones that were identified as either induced or repressed by RA were analysed quantitatively.
The time course and extent of induction of B-galactosidase activity was assessed for each cell line using a quantitative assay. ES cells (2x10 high5) were plated onto 35mm gelatinised tissue culture dish in ES cell medium containing 15% FCS and LIF. After overnight culture, and then every 6 hours subsequently, the medium was changed on all plates such that cells were in the continuous presence of RA for varying time periods ( 6, 12,24 or 48 hours). Control samples were in ES cell medium containing 5% FCS throughout the experiment. Cells were harvested by trypsinisation, washed in PES and resuspended in 100�l 0.25M Tris pH 7.5. After 3 freeze/thaw cycles, samples were centrifuged at 13000rpm for 5 mins and the protein concentrations in the supernatant determined. The B-galactosidase assays were performed using equivalent amounts of protein (75-200�g) in 0.5ml Bgal buffer (60mM Na2HPO4, 40mM NaH2PO4, 10mM KCL, ImM MgC12, 5mM Emercaptoethanol) containing 0.2mg O-Nitrophenyl E-D-Galactosidase. Incubation was carried out overnight and the optical density at 420nM was determined.
Isolation of RA-responsive cell lines. 6.5 x 10 high7 ES cells were electroporated with the PT-l ATG vector and 3600 ES cell colonies were replica-plated, induced with RA and stained for B-galactosidase activity. 202 B-galactosidase-positive colonies were picked from the master plate into 24 well plates, expanded and lacZ expression retested in the presence and absence of RA. 9 colonies were identified in which the reporter gene was induced by RA and 11 that were repressed.
Table 1 shows the quantitative induction of E-galactosidase activity in the RA-responsive cell lines at various times after exposure to RA. The results are expressed as a fold induction compared to the control sample (0) that was not treated with RA. The induced lines varied greatly in the level of induction and in the time course over which the induction was observed. For example, the level of reporter gene expression in ES cell lines 1.23,1.75 and 1.114 was induced two-fold after 48 hours, whereas in line 1.210, over the same time period, [:)-gal activity was induced 16 Table 1. Results of quantitative B-galactosidase
assays on RA-responsive genetrap ES cell lines expressed 
We have made chimeric embryos with some of the RA-responsive cell lines and preliminary results suggest that a high proportion (> 70%) of the RA-responsive genes that have been trapped are expressed in a spatially and temporally restricted manner during embryogenesis. Further work is being done to confirm this result.
We describe the development of a gene trap screen in ES cells which pre-selects in vitro for integrations into genes that lie downstream of ligand/receptor-mediated signalling pathway. RA was used to test the general applicability of this method. We have successfully isolated 20 ES cell lines which carry gene trap vector integrations into RA responsive genes. The induction protocol was designed to identify genes that were either directly or indirectly responsive to RA. Gene trap lines were treated with RA for 48 hours to identify indirectly responsive genes (ie genes that are regulated as a consequence of RA-induced differentiation). We also added fresh RA-containing medium 6 hours prior to staining to identify genes that were possibly induced at an earlier time point and thus candidates as direct genes. The time courses of induction of the isolated lines suggest that the trapped, endogenous genes are not directly responsive to RA but rather lie further downstream in the RA-induced differentiation pathway. Gene that are directly induced by RA are normally induced within 6 hours and do not require protein synthesis for induction. However, the promoter region of the laminin B1 gene contains a retinoic acid binding element (RARE) but the induction of expression of this gene is not seen until 26 hours after RA treatment (Vasios et al, 1991). Obviously, the mode of RA regulation of the trapped gene can only be accurately assessed by a detailed analysis of their transcriptional regulatory regions. We predict that a large proportion of these RA-responsive genes will be developmentally-regulated genes and our preliminary analysis supports this. We have introduced several of these ES cell lines into mouse embryos and have very specific temporal and spatial lacZ expression patterns. This approach could be adapted to screen for genes that act downstream of any ligand-mediated pathway such as polypeptide growth factors. The success of this will be dependent on the expression of the appropriate receptors in ES cells but may be complicated by the autocrine expression of the growth factor in ES cell cultures. As ES cells can differentiate into haematopoietic lineages in vitra (Doetschman et al., 1985, Wiles & Keller, 19) it may be possible to directly select gene trap integrations into genes that are expressed in haematopoietic lineages using this type of directed approach.
Beddington, R.S.P. & Robertson, E.]. (1989) An assessment of the developmental potential of embryonic stem cells in the midgestation mouse embryo. Development 105: 733-737. Conlon, R.A. & Rossant, ]. ( 1992) Exogenous retinoic acid rapidly induces anterior ectopic expression of murine Hax-2 genes in viva. Development 116: 357-368. Doetschman, T., Eistetter, H., Katz, M., Schmidt, W. & Kemler, R. (1985). The in vitra development of blastocyst-derived embryonic stem cell lines: formation of visceral yolk sac, blood islands and myocardium. ]. Embryol. exp. Morph. 87:27-45. Eichele, G. ( 1989) Retinoids: signalling molecules in vetrebrate pattern formation. Trends Genet. 5: 246-251. Evans, M.]. & Kaufman, M.H. (1981) Establishment in culture of pluripotential cells from the mouse. Nature 292: 154-156. Freidrich, G. & Soriano, P. ( 1991) Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes & Dev. 5: 1513. Gossler, A., Joyner, A., Rossant, ]. & Skarnes, W.C. (1989) Mouse embryonic stem cells and reporter constructs to detect developmentally regulated genes. Science 244: 463-465. Hill & Wurst, W. (1993) Screening for Novel Pattern Formation Genes Using Gene Trap Approaches. Methods in Enzymology 225: 664-681. Nagy, A., Rossant, ]., Nagy, R., Abramow-Newerly, W. & Roder, ].C. (1993). Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc. Natl. Acad. Sci. USA 90: 8424-8428. Simeone, A., Acampora, D., Arciono, L., Andrews, E., Boncinelli, E. & Mavilio, F. ( 1990) Sequential activation of HOX2 homeobox genes by retinoic acid in human embryonal carcinoma cells. Nature 346: 763- 766. Simeone, A. Acampora, D., Nigoro, V., Faiella, A., D'Esposito, M., Stornaiuolo, A., Mavilio, F. & Boncinelli, E. ( 1991) Differential regulation by retinoic acid of the homeobox genes of the four HOX loci in human carcinoma cells. Mech. Dev. 33: 215-228. Skarnes, W.C. (1993). The identification of new genes: Gene trapping in Transgeneic Mice.. Current Opinion in Biotechnology 4:684-689. Skarnes, W.C., Auerbach, A.B. & ]oyner, A.L. (1992). A gene trap approach in mouse embryonic stem cells: the lacZ reporter is activated by splicing, reflects endogenous gene expression, and is mutagenic in mice. Genes Dev. 6: 903-918. Vasios, G., Mader, S., Gold, ].D., Leid, M., Lutz, Y., Gaub, M-P., Chambon, P. & Gudas, L.]. ( 1991) The late retinoic acid induction of laminin B1 gene transcription involves RAR binding to the responsive element. EMBO ]. 10: 1149-1158. Wiles, M.V. & Keller, G. (1991) Multiple haematopoietic lineages develop from embryonic stem (ES) cells in culture. Development 111: 259-267. Wurst, W. & ]oyner, A.L. (1993). Production of targeted embryonic stem cell clones. In: Gene Targeting, A Practical Approach. ed. A.L.Joyner, IRL Press, Oxford. Wurst, W., Rossant, ]., Prideaux, V., Kownacka, M., ]oyner, A.L., Hill, D.P., Guillemot, F., Gasca, S., Auerbach, A. & Ang, S-L. (1994). An embryonic expression screen for spatially restricted genes using a gene trap approach in ES cell chimeras. Genetics (in the press) . |