Hämatol. Bluttransf. Vol 35 |
|
1 Program Resources/DynCorp, Frederick Cancer Research
and Development Center, Frederick, MD 21702-1201, USA.
Cystic fibrosis (CF) is an autosomal recessive disorder, and di Sant' Agnese et al. [3] found that the sweat of CF patients contains an excess of sodium and chloride ions. Defects in the regulation of chloride ion transport have been documented in CF epithelial cells [5, 10, 14, 16]. The chloride channel normally responds to ßadrenergic agents, but CF cells are defective in this response [6,9, 14]. It has been proposed that the CF defect involves a pathway whereby cAMP regulates ion transport. The symptoms of CF patients are heterogeneous between and within families [13]. Although most individuals are diagnosed by the time they reach the age of ten, a few remain undiagnosed until adulthood [2, 15]. Approximately 15% of CF patients do not require supplemental pancreatic enzymes and are designated as pancreatic sufficient (PS) [7]. PS is typically concordant within families, suggesting that PS patients may have less severe mutations in the CF gene. However, the heterogeneity within families suggests that additional genetic and environmental factors contribute to the severity of the disease. The molecular cloning of the CF gene has provided additional research strategies to further understand the disease, and the regulation of ion transport in secretory cells. The gene encodes a 170 kDa polypeptide that is a member of a superfamily of membrane-bound active transport molecules [4, 11, 12]. A threenucleotide deletion in a putative A TP binding domain has been found in 70 % of CF chromosomes; this alteration removes a phenylalanine codon at position 508 (delta F 508). To further understand the relationship between mutations in the gene and the phenotype of patients, we have examined a group of patients who do not contain the common mutation on both chromosomes.
To identify mutations in the CF gene, specific regions were amplified by the polymerase chain reaction (PCR) and assayed for single-stranded conformation polymorphisms (SSCPs). This newly described method allows the rapid screening of samples for the presence of genetic variation [8]. SSCPs are detected by denaturing the DNA and resolving it on nondenaturing acrylamide gels. Each strand of the DNA fragment can potentially form a unique conformation (and have a distinct mobility), and any mutation within that segment can potentially affect the mobility. Table I. Detection of mutations using
the SSCP technique 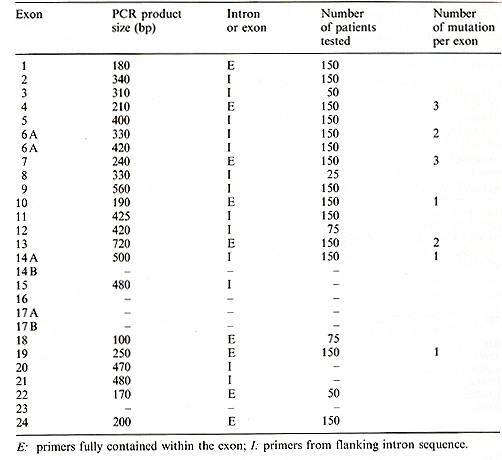
Table 2. CFTR mutations 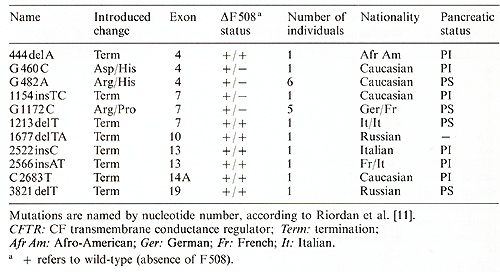 
Because the most common mutation accounts for only 70% of CF chromosomes [4], a large proportion of CF patients (40 % -50% ) are compound heterozygotes, i.e., they have two different mutations in the gene. Thus there is a large number of possible genotypes found in CF patients. This appears to account, in part, for the variation observed in the phenotype of patients. However, within families affected individuals can show differences in sweat chloride levels and severity, demonstrating that additional genetic and/or environmental factors contribute to these phenotypes. The clearest correlation between the patient's genotype and phenotype is seen in the pancreas. All patients we have observed that are homozygous for the delta F 508 deletion are PI. However, even in these patients, genetically identical at the CF locus, there is considerable variation in clinical outcome. This variation is expressed in the age of diagnosis, pulmonary function, and sweat chloride value. In the lungs of CF patients, damage is principally caused by bacterial infection. These infections are believed to be secondary to the abnormal mucus present in patients. Furthermore, immune function genes such as the human lymphocyte antigens (HLA) and/or the T cell receptor locus could playa role in the susceptibility and/or response to bacterial infection.
We thank McNeil Pharmaceuticals for support of research in the USSR. Marga Belle White is supported by a postdoctoral fellowship from the US Cystic Fibrosis Foundation. This project has been funded at least in part with Federal funds from the Department of Health and Human Services under con tract n um ber N O 1-CO- 74102 with Program Resources, Inc. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
1. Dean M, White MB, Amos J, Gerrard B, Stewart C, Khaw K- T, Leppert M (1990) Multiple mutations in highly conserved residucs are found in mildly affcctcd cystic fibrosis patients. Cell 61 : 863- 870 2. Di Sant' Agnese PA, Davis PB ( 1979) Cystic fibrosis in adults. Am J Med 66:121-132 3. Di Sant' Agnese PA, Darling RC, Perea GA, Shea BA (1953) Abnormal electrocyte composition of sweat in cystic fIbrosis of the pancreas: clinical significance and relationship to the disease. Pediatrics 12:549-563 4. Kerem B-S, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui L-C (1989) IdentifIcation of the cystic fibrosis gene: genetic analysis. Science 245: 1073- 1080 5. Knowles M, Gatzy J, 13oucher R (1981) Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Engl J Med 305:1489-1495 6. Knowles MR, Stutts MJ, Spock A, Fischer N, Gatzy JT, Boucher RC (1983) Abnormal ion permeation through cystic fibrosis respiratory epithelium. Science 221:1067-1069 7. Kopelman H, Durie P, Gaskin K, Weizman Z, Forstner G (1985) Pancreatic fluid secretion and protein hyperconcentration in cystic fibrosis. N Engl J Med 312: 329 334 8. Orita M, Suzuki Y, Sekiya T, Hayashi K (1989) Rapid and sensitive detection of point mutations and DNA polymorphisms using the polymerase chain reaction. Genomics 5: 874- 879 9. Quinton PM (1983) Chloride impermeability in cystic fibrosis. Nature 301 : 421-422 10. Quinton PM, Bijman J (1983) Higher bioelectric potentials due to decreased chloride adsorption in the sweat glands of patients with cystic fibrosis. N Engl J Med 308:1185-1189 11. Riordan JR, Rommens JM, Kerem B-S, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou J-L, Drumm ML, Iannuzzi MC, Collins FS, Tsui L-C (1989) Identification of the cys tic fibrosis gene: cloning and characterization of complementary DNA. Science 245:1066-1073 12. Rommens JM, Iannuzzi MC, Kerem B-S, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, Zsiga M, Buchwald M, Riordan JR, Tsui L-C, Collins FS (1989) Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 245: 1059 -1065 13. Rosenstein BJ, Langbaum TS (1984) Diagnosis. In: Taussig LM (ed) Cystic fibrosis. Thieme-Stratton, New York, pp 87-114 14. Sato K, Sato F (1984) Defective beta adrenergic response of cystic fibrosis sweat glands in vivo and in vitro. J Clin Invest 73:1763-1771 15. Su CT, Beanblossom B (1989) Typical cystic fibrosis in an elderly woman. Am J Med 86: 701- 703 16. Welsh MJ, Liedtke CM (1986) Chloride and potassium channels in cystic fibrosis airway epithelia. Nature 322:467-470 17. White M, Amos J, Hsu JM-C, Gerrard B, Finn P, Dean M (1990) A frameshift mutation in the cystic fibrosis gene. Nature 344:665-667 |