|
* This work was generously supported by the Deutsche Forschungsgemeinschaft (SFB 47 "Virologie", 103 "Zellenergetik und Zelldifferenzierung", 118 "Früherkennung des Krebses"), by the Bundesminister fur Forschung und Technologie, by the President of the University of Giessen, 6300 Giessen, FRG, and by the President of the University of Erlangen, 8520 Erlangen, FRG The present paper supplements that of "The Biology of an Oncogene" published in vol. 28 of this series [ I] A. Introduction Currently in cancer research much emphasis is being placed on mutation [2-4], rearrangement [5], amplification [6-8], and demethylation [9] of oncogenes that are supposed to represent the primary events leading to oncogene activation when carcinogens trigger neoplasia in animals or humans (the reader is referred to ref. [10] for more extensive details on this topic). The data underlying this supposition were mainly derived from experiments performed with tumor cells in vitro. In vivo studies on this issue are rare. A model which has proven to be suitable for such in vivo studies is Xiphophorus, i.e., a fish genus from Central America known as the platyfish and swordtails [ II ]. All individuals of this genus carry an oncogene, designated Tu, which was analyzed by formal genetics long before the viral and cellular oncogenes were identified by methods of modern molecular biology [ l2 -16]. Our phenogenetic, cytogenetic, and developmental genetic studies on Xiphophorus, however, have failed so far to detect any structural alterations of the Tu oncogene which were expected to occur when the gene switches over from the silent to the tumorigenic state. In contrast, it appears that impairment of the Tu oncogene might reduce rather than promote its tumorigenic potential and, therefore, it is difficult to detect. By means of seven experiments the present study aims to show that the common primary events in the causation of neoplasia in Xiphophorus are changes in the regulatory gene systems controlling the oncogenes rather than changes in the oncogenes themselves.
I. The Oncogene
I. First Experiment: Tumors Induced by Dislocation of Regulatory
Genes for the Oncogene in a Germ Line Cell 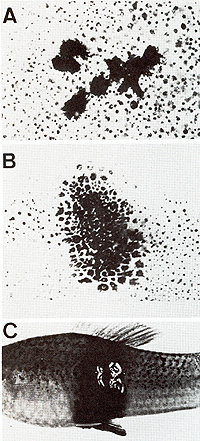 Fig.1A-C. Development of a somatic mutationconditioned melanoma. A Cell clone consisting of eight transformed melanocytes; B Clone consisting of some hundreds of transformed melanoblasts and melanocytes; C Lethal malignant melanoma 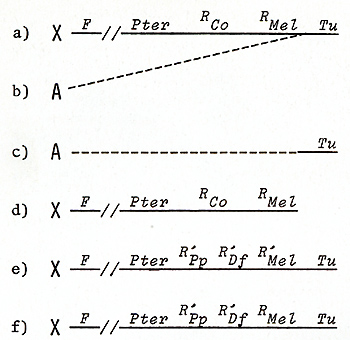 Fig. 2 a-f. a-c Translocation of the Tu oncogene from the X chromosome (X) of x. maculatus to an autosome (A) of X. helleri and d the respective Tu deletion. Note separation of Tu from its linked regulatory genes RMel and Rco (see first experiment of this study); e X chromosome of X. maculatus that is used in the second experiment (see Figs. 5, 9); f The X chromosome used in the third experiment (see Fig. 10). Note that e and f differ only in the RMel locus. F, female-sex-determining region; Pter, pterinophore locus. Rco region containing at least 13 compartment-specific regulatory genes such as Rpp (posterior part-specific) and RDf (dorsal fin-specific); RMel, melanophore-specific regulatory gene; Tu, oncogene neoplastic transformation takes place in the pigment-cell precursors
of the early embryo. In the segregants lacking both the linked and
the nonlinked regulatory genes, neoplastic transformation continues
in pigment-cell precursors in all areas of the body, thus forming
a "whole body melanoma " (Fig. 3 B) which kills the fish before
or shortly after birth. If, however, the nonlinked regulatory gene
is present in the system, melanoma development is retarded at the
time of birth (Fig. 3 C). The fish develop melanoma but may reach
sexual maturity, thus providing the possibility to breed by further
backcrosses with x. helleri, a stock that continuously produces
lethal-melanoma-developing and nonlethal-melanoma-developing embryos.
If, furthermore, the regulatory gene is introduced into the system
in the homo zygous state, no Tu-mediated melanoma develops (Fig.
3 A). Fish carrying the Tu deletion chromosome (see Fig. 2 d) have
a highly diminished potential for the formation of all kinds of
neoplasms. The development of the whole body melanoma in the early
embryo reflects the genuine oncogenic effect of the com pletely
deregulated Tu on the pigment-cell system. These observations suggest
to us that in the early embryo Tu normally exerts important functions
in cytodifferentiation, which, if Tu is completely deregulated,
cannot be stopped (see below). So far we have not tested pp60c-src
kinase activity in the embryos that develop melanoma. However, pp60c-src
kinase activity was measured in the normal developing embryos (Fig.
4) [21]. The activity is detectable from the very outset of cleavage
and strongly increases during early organogenesis. While organogenesis
morphologically culminates, kinase activity apparently becomes choked,
decreases during the growth phase before and after birth to a lower
level, and thereafter (not shown in Fig.4) remains constant at a
basic level throughout the whole life of the fish [ 19]. In adults
it was found that pp60c-src kinase activity is organ specific, with
the brain always showing the highest values [ 18, 19]. The results
suggest that c-src activity is related to differentiation and specificity
of organs rather than to cell growth. Similar sequences of pp60c-src
kinase activity during life were found in frogs and chickens (see
Fig. 4), indicating that our results are more general rather than
fish specific [21]. These genetic, embryological, and molecular
biological observations suggest to us that the melanoma-mediating
oncogene, irrespective ofits appearance as Tu or c-src, exerts important
normal functions as a developmental gene in the early embryo. Moreover,
we assume that in normal embryogenesis these functions become switched
off or choked by regulatory genes after cell differen tia ti on
progresses to organogenesis. If, however, the regulatory genes (i.e.
the entire choke in the lethal Tu translocation) are lacking, the
oncogene continues to exert its early embryo-specific functions
which, as an extension of the cellular development in the early
embryo, 
appear as neoplastic transformation. Support for this idea comes from the high pp60c-src kinase activity that is found in the melanoma of young and adult fish (see second and third experiments). In any case, the crucial event leading to neoplasia in this experiment is the X-ray-induced translocation -condi tioned loss of oncogenespecific regulatory genes.
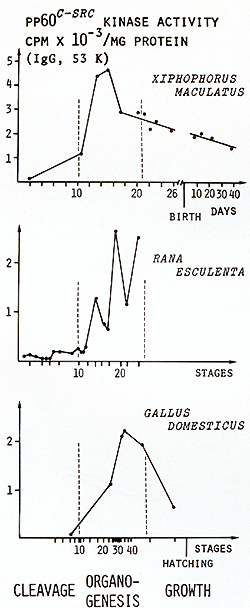 Fig.4. pp60c-src Kinase activity during cleavage, organogenesis, and growth. Da ta from [21 ] Backcrosses of the fish carrying the malignant melanoma with the
swordtail show a different result: 50% of the BC segregants develop
malignant melanoma, whereas the remaining 50% are melanoma free;
benign melanomas do not occur. Whenever melanomas occur in these
crossing experiments, they develop in both the compartment of the
dorsal fin and the compartment of the posterior part of the body.
The results obtained in this crossing experiment (and a large variety
of similar experiments) may be explained by the assumption of four
prominent genetic components that are involved in melanoma formatioll
or protection from melanoma, respectively. They are contributed
by x. maculatus (for those contributed by x. helleri but not involved
in melanoma formation in this crossing experiment, see below) and
are normal homozygous constituents of the natural gene pool of the
wild platyfish population from Rio Jamapa. Three of the genetic
components (see the following points 1, 2, 3), as was confirmed
by about 70 structural changes, are closely linked together at the
end of the X chromosome (see Fig. 2). The fourth (point 4) is autosomally
located. 
4. The differentiation gene Diff This gene is indicated by the I: I segregation of benign- and malignant-melanoma-bearing fish. If it is present, the majority of the melanoma cells are well differentiated (benign melanoma). If, however, Diff is lacking, the majority of the melanoma cells are poorly differentiated (malignant melanoma ). Additional regulatory genes contributed by the platyfish to the
hybrid genome have been identified but are considered only in general
in this experiment. Xiphophorus helleri also contains the Tu oncogene
and presumably the linked regulatory genes, which, however, are
not mutated and thus are fully active. Tu and its linked regulatory
genes, therefore, are not detectable with the methods used in this
experiment. Furthermore, since the linked regulatory genes act only
in the cis position, this "oncogene-regulatory-gene complex," contributed
by the swordtail, does not significantly influence the expression
of the platyfish-derived melanoma-mediating Tu copy. No Diffand
nor any other nonlinked regulatory gene was found in the swordtail
that might correspond to those of the platyfish. The outcome of
this crossing experiment suggests the following interpretation:
Following crossings and backcrossings according to Fig. 5, the regulatory-gene-carrying
chromosomes of the platyfish are replaced stepwise by the homologous
chromosomes of the swordtail, resulting in the gradual disintegration
of the regulatory gene system for the platyfish-derived Tu. Thus
the Tu hybrids spontaneously develop benign melanoma if some regulatory
genes such as Diff are still present in the genome and malignant
melanoma if the regulatory genes are lacking. If the platyfish-derived
Tu is lacking, no melanomas occur . Based on the assumption that
the activity of the pp60c-src-associated phosphokinase monitors
the activity of the c-src oncogene, we carried out comparisons between
kinase activity, i.e., c-src expression, and tumor development,
i.e., Tu expression. The preparatory work to this study showed that
pp60c--src kinase activity in normal tissues of non tumorous and
tumorous fish is tissue specific, with the brain always showing
the highest values [ 18]. It furthermore showed that this activity
is elevated in the brain of benign-melanoma-bearing fish, and even
higher in the brain of malignant-melanoma-bearing fish. In any case,
pp60c--s'rc kinase activity varies in both melanoma and the brain
in the same direction. Hence, we were able to determine pp60c-src-associated
protein kinase activity mainly in brain extracts and relate the
activity observed to the expression of Tu ascertained by the development
of melanoma. The possibility that the differences in kinase activity
measured in the fish of different Tu genotypes are due to secondary
processes in the melanoma appears unlikely. Therefore, the results
reflect the actual genetic activity of the c-src oncogene in the
nontumorous brain tissue of the tumorous and nontumorous fish. For
critical evaluation of the methods see refs. [18, 19]. As indicated
in Fig. 5, the purebred platyfish female (Fig. 5 A) carrying the
population-specific "Tu-regulatory-gene complex" in both of its
X chromosomes, as well as the purebred swordtail and the BC hybrids
lacking this special complex, display the same activity of c-src
kinase. We interpret this activity to be the basic expression of
c-src. In contrast, the melanomabearing hybrids which contain the
derepressed Tu show an increase in c-src activity, with the malignant-melanoma-bearing
BC hybrids displaying the highest activities. To accomplish the
crossing experiment (not shown in Fig. 5) we crossed two melanoma-bearing
BC hybrids (FX F) that exceptionally reached sexual maturity. The
resulting Tu/Tu hybrids (25% of the offspring) are of special in
terest. They show an oncogene dosage effect in both melanoma forma
tion and pp60c-.src kinase activity as compared with the parental
BC hybrids [1, 20]. In summary, all experiments of this kind showed
a clear-cut correlation between c-src expression (measured by pp60c--s'rc
kinase activity) and Tu expression (measured by melanoma formation).
Many features that are assumed to be .involved in the causation
of neoplasia have been found in melanoma of Xiphophorus. About 70%
of the mel'anomatous cells in cell cultures derived from embryos
exhibit at least two pairs of double minute chromosomes (Fig. 6)
[22) that might be interpreted 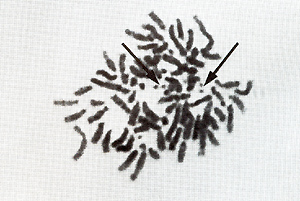 Fig.6. Metaphase showing double minute chromosomes (DM). OMs were always found in cell cultures derived from melanomatous cmbryos [22] 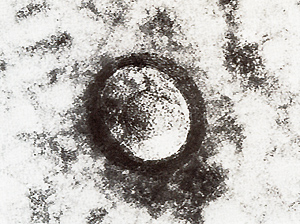 Fig. 7. Virus particle (iridovirus-like [24]). These particles were always found in the supernatant of cell cultures mentioned in the legend of Fig.6 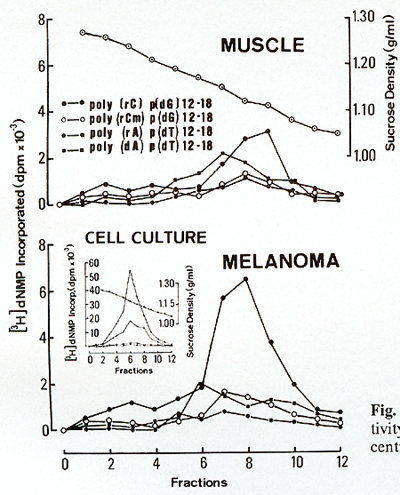 Fig.8. RNA-dependent DNA polymerase activity in Xiphophorus.
as carriers of amplified oncogenes [7, 8]. Virus particles have been found in the melanoma following treatment with bromodesoxyuridine [23]. Following sucrose density centrifugation, in the supernatant of nontreated melanomatous cells of cultures mentioned above, RNA-dependent DNA polymerase associated with particles similar to fish iridovirus [24] (Fig. 7) was found in the density range of 1.15-1.17 g/ml. Three to five particles have been counted per grid, which is equivalent to 105 particles/ml. RNA-dependent DNA polymerase activity has also been detected in the melanoma tissue and, although three to five times lower, in the muscle (Fig. 8). Activity of many other enzymes, such as tyrosinase [25], lactate dehydrogenase [26, 27], malate dehydrogenase, and pyruvate kinase [27], is changed in the melanoma. Table 1. SCE frequency in intestinal cells of Xiphophorus
[29] 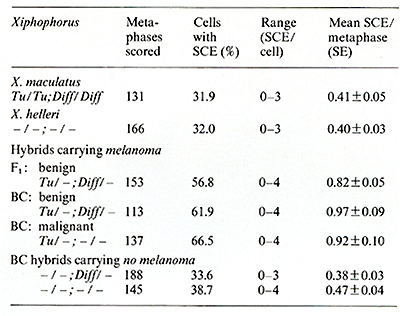 Sister chromatid exchange (SCE) is elevated in melanoma cells [28]. Interestingly, its high frequency is in parallel with that of non tumorous intestinal cells of melanomatous fish, and is low in intestine of the nonmelanomatous fish (Table 1) [29]. This is a striking similarity to the parall.els of pp60c-src kinase activity measur~d In the melanoma and in the healthy braIn. These factors and many others not mentioned in this study are certainly important in melanoma formation, but probably none of them is involved in the primary event of tumorigenesis in this experiment. The primary event of tumor initiation in this experiment, without any doubt, happens when the egg and sperm contrIbute a normal set of oncogenes to the zygote but fall to contribute the adequate regulatory gene systems for control of the oncogenes. The confirmation of this view comes from the final crossing series of this experiment outlined in Fig. 9: Backcrosses of malignantmelanoma-bearing hybrids (Fig. 9 A) (i.e., Fig. 5 F) with the platyfish (Fig. 9 B) (like those of Fig. 5 A, but containing the Tu deletion according to Fig. 2 d) result in a quasi Fl that segregates in 50% of animals displaying benign melanoma (Fig. 9 C) and 50% exhibiting neither melanomas nor spots (not shown in Fig. 9). Further backcrosses of the benign melanoma bearing quasi-Fl hybrids with the platyfish 
(Fig. 9 D) result in spotted and nonspotted fish that are similar
to the purebred platyfish (Fig. 9 E-H). Genetically they segregate
into those that inherit the capability to develop melanoma after
crossings .with swordtails and those that do not Inherit the capability
to develop melanoma (not shown in Fig. 9). The outcome of this final
series of backcrosses indicates the step wise reintroduction of
the platyfish chromosomes carrying the regulatory genes for the
oncogene into the descendants, and the reconstruction of the original
regulatory gene system that suppresses the activity of the oncogene
in the platyfish genome. 
Presumably the oncogene itself remained unchanged during the crossing procedure (Figs. 5, 9) that first induced and thereafter suppressed melanoma formation. Carcinogens or tumor-suppressing agents were not required in this experiment. The outcome of the experiment, however, indicates that the experimenter must interfere in the regulatory gene system of the oncogene to be able to induce or suppress melanoma ( or any other neoplasm). To examine this idea, we carried out the following experiment. This experiment is based on the two assumptions that (a) the total number of pigment-cell precursors competent for neoplastic transformation is 10 high 6 (this is the average number in the pigment-cell system of a young fish) and (b) the mutation rate for a given gene in these cells is 10 high-6. Then tumor incidence is one ( on average 100% of the treated animals develop one tumor) provided the oncogene is under control of only one regulatory gene. If, however, the oncogene is regulated by two genes, the rate of simultaneous mutations of both regulatory genes in one cell is 10high-12 and the tumor incidence is 10high-6. This calculation shows that if the oncogene is controlled by more than one regulatory gene, it is difficult to succeed in inducing somatic mutation-conditioned neoplasms at a frequency suitable for mutagenesis experiments. To establish a Xiphophorus strain suitable for mutagenesis studies on a single regulatory gene of the Tu oncogene, we replaced the R`Pp RDf R'Mel platyfish X chromosome used so tar (Fig. 2 e) by the Rpp RDj R Mel Tu X chromosome (Fig. 2 f), which, instead of the impaired RMel, contains the normal RMel. Crossings were performed according to Fig. 10, which are essentially the same as those described in the first part of the previous experiment (Fig. 5). Due to the active RMel that controls the oncogene in the pigment cell system, no melanomas develop in the hybrids. The BC hybrids E and F that correspond to the melanoma-developing BC hybrids in the former experiment (they can be recognized by a reddish coloration encoded in the pteridine marker gene PIer also linked to the Tu oncogene; see Fig. 2) are, however , sensitive to carcinogens and may develop Table 2. Tumor initiation (somatic mutation)
and promotion (promotion of cell differentiation) distinguished
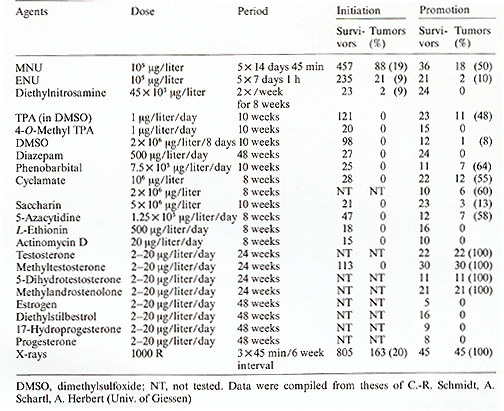
Table 3. Elevated pp60c-src kinase activity
in Xiphophorus bearing tumors 
To perform this transfection experiment we used different laboratory
stocks of x. maculatus as donors. These stocks were uniform with
respect to Tu and the impairment of the compartment-specific regulatory
genes RAp and Rpp (see fifth experiment). They were, however, different
in the condition of the melanophore-specific regulatory gene RMeI,
which was normal, impaired, strongly impaired, or deleted (Fig.
12). Total genomic DNA of the donors was injected into the neural
crest region of early embryos (where the pigment-cell precursors
originate) of x. helleri. This species has excellent potential for
expressing a deregulated oncogene (see previous experiments). If
a donor containing the normal RMeI-Tu complex (Fig. 12 A) was used,
no recipient exhibited transformed pigment cells. If a donor strain
was used that carried a Tu slightly derepressed in the pigment-cell
system by a "weak" mutation of its linked RMeI, i.e., R'Mel Tu (Fig.
12 B), then 0.4% of the recipients developed colonies of neoplastically
transformed cells. If the DNA originated from a strain that carries
the Tu derepressed to a greater degree, due to a stronger mutation
of RMel. i.e. R``MeI (Fig. 12 C), the incidence of recipients exhibiting
transformed pigment cells increased to 2.6%. If finally, a donor
was used in which the Tu lacks the RMeI (due to a chromosomal translocation)
( Fig. 12 D), this incidence increased to 6.3%. DNA from animals
carrying additional but rigidly repressed accessory Tu copies (not
shown in Fig. 12) did not influence the incidence of transformants
[34]. Besides the fact that the information for neoplastic transformation,
presumably the 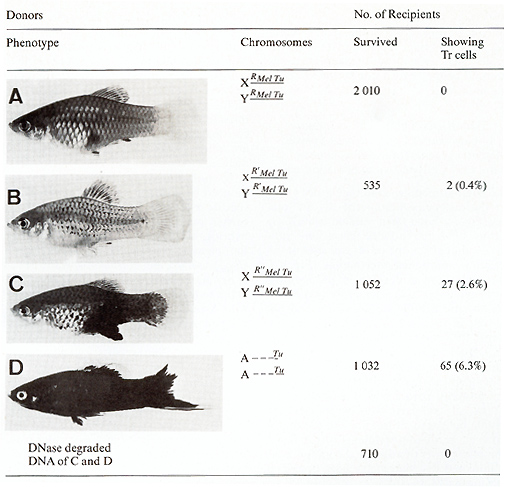 Fig.12A-D. Transfection activity of donor DNA extracted from male gonads of fish differing in Tu control by the pigment-cell-specific regulatory gene R Mel. Data from [34 ]. See text oncogene itself, was transferred via total genomic DNA, it is important to note that the transforming donor DNA did not originate from tumor cells but from the nonneoplastic testes, indicating that oncogenes must not necessarily be changed or amplified in order to acquire the transforming potential. The many oncogene transfection experiments accomplished during the past years by several authors with other systems in which DNA extracts from tumors were used [2, 3, 35-37], with the expectation that tumor DNA differs from DNA from normal tissues, should be, in our opinion, reconsidered from the viewpoint of repression and derepression of oncogenes exerted by intact and defective regulatory genes. The main factors responsible for neoplasia are, in view of our results on Xiphophorus, not the onc genes, but their regulatory genes. This view is supported by the fact that the incidence of transformants, i.e., the incidence of transformation events mediated by Tu, was independent of the number of Tu copies in the donor DNA (we tested DNA containing up to eight copies), but was exclusively dependent on the degree of impairment of RMel. In this light it also appears reasonable to assume that R Mel, if present, is so closely linked to Tu that both Tu and RMel are always cotransferred [34]. Although the donor DNA originated from fish exhibiting different degrees of Tu expression, the transformed cells of the recipients all looked alike, and the cell colonies were all about the same size. This indicates that growth of the tumor is influenced neither by Tu nor by the intact or impaired RMel.
The compartment-specific regulatory genes for the oncogene, designated Rco in total (see Fig.2), have been studied mainly by means of X-ray-induced germ line mutations that affect one or several sites of the crossing-conditioned melanomas (Fig. 13). These melanomas develop in BC hybrids, for instance (a) in the dorsal fin; (b) in the tail fin; ( c) in both the dorsal fin and tail fin; (d) in the anal fin; (e) in the tail fin, dorsal fin, anal fin, mouth tip, and posterior part of the side of the body (mutations of five compartment-specific regulatory genes are involved); (f) in the anterior and posterior parts of the side of the body; (g) in all compartments except for the mouth, belly, eye, dorsal fin, and tail fin; or (h) even in all compartments of the body. The phenotypes of additional combinations of impaired Rco genes have been described previously [ 13, 15, 16]. The compartment-specific distribution of these melanomas is inherited according to the segregation of the parental Tu-carrying chromosome, indicating that the respective Rco genes are linked to Tu, and structural changes of the chromosome have verified that this linkage is very close. At least 14 genes corresponding to 14 different compartments have been identified. They represent regulatory genes that were designated RAp (anterior part), Rpp (posterior part), RDf(dorsal fin), RTf(tail fin), etc. in reference to each specific body compartment. Intact Rco genes repress Tu, and impaired Rco genes permit Tu activity. They act in the cis position only, indicating that the compartment-specific regulation of Tu exerted by the Rco genes acts at the DNA level. In the active state the Rco genes appear to delay the differentiation of pigment cells in the stem-cell stage (S-melanoblasts; see Rco in Fig. 14). Additional mechanisms that are not understood provide the fish with differentiating pigment cells that mostly escape neoplastic transformation, but very exceptionally may be transformed. If, however, one or several R Co genes are impaired by mutation, compartment-specific melanomas develop in the hybrids and are inherited according to Mendelian prediction. To study whether melanoma can also be induced by impairment of a compartmentspecific regulatory gene in a somatic cell we carried out the following experiment: BC hybrids, which, due to the impairment of the pigment-cell-specific regulatory gene (R`Mel) and of the dorsal-fin-specific regulatory gene (R`Df), develop hereditary melanoma in the dorsal fin only, were irradiated with X-rays. These animals frequently developed additional melanomas in other compartments of the body (Fig. 15) which, in contrast to the hereditary melanoma of the dorsal fin, are nonhereditary and develop from small foci of transformed cells in the skin. This indicates a mutational event in a particular compartment-specific regulatory gene in a particular competent pigment-cell precursor in a particular compartment of the body. The oncogene itself that mediates neoplastic transformation probably remained unchanged.
The neutral crest cells of Xiphophorus which are, like those of
all vertebrates, the precursors of different cell types, start migrating
and differentiating at the outset of organogenesis in the embryo
(Fig. 14) [38, 39]. Those entering their final locations in the
skin and extracutaneous tissues become determined to differentiate
to chromatoblasts. These chromatoblasts are the common precursors
of all types of pigment cells (chromatophores), including pterinophores,
purinophores, and melanophores. Those chromatoblasts committed to
differentiate to melanophores give rise to stern melanoblasts (S-melanoblasts).
These may reproduce throughout the life of the fish,  Fig.13. Compartments for the development of melanomas in the fish. Mo, mouth; Ey, eye; Bm. brain membrane; Ap, anterior part; Df dorsal fin; Pt. peritoneum; Af anal tin; Sc, spinal cord; Pp, posterior part; Cr, crescent region; Pd. peduncle of the tail fin; Cf: caudal tin stripe; Tf tail tin (see text) 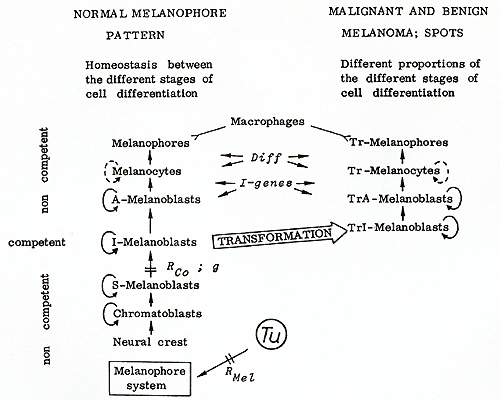 Fig. 14. Schematic presentation of the differentiation of normal and neoplastically transformed pigment cells. S -, I -, and A -melanoblasts are stem, intermediate, and advanced melanoblasts, respectively. The Tr cells represent the transformed cells. Only J-melanoblasts are competent for neoplastic transformation. Tu, tumor gene (oncogene); RMel, regulatory gene for control of Tu in the melanophore system; Rco, compartment-specific regulatory genes; g, "golden" gene that blocks pigment-cell differentiation; Diff, differentiation gene; I -genes, intensifier genes, which support proliferation of poorly differentiated transformed pigment cells. Macrophages attack melanophores and Tr-melanophores. Modified from [ 16] but may also differentiate to intermediatestage melanoblasts (l-melanoblasts)
that continue differentiation to the advancedstage melanoblasts
(A-melanoblasts) that can be distinguished from their precursors
by their reaction to dopa. These cells differentiate to melanocytes,
and these, finally, to melanophores. Genetic, cytological, and ultrastructural
studies of the differentiation of the pigment cells have shown that
the l-melanoblasts are the only cells of the system that are competent
to undergo neoplastic transformation, i.e., corn petent to undergo
the transforming activity of the Tu oncogene [38, 39]. The I-melanoblasts,
after being transformed to Trl-melanoblasts (all transformed cells
are called Tr cells) differentiate to the easily recognizable, proliferating
TrA melanoblasts. These Tr cells differentiate to the heavily pigmented
Tr melanocytes, which proceed to the terminal stage of differentiation
of the transformed pigment cells, represented by the Tr melanophores
[15]. We have analyzed several genes that are involved in pigment-cell
differentiation. One of these genes is the "golden " gene (g+),
which, if present as a mutation in the homozygous state (g/ g),
creates an almost complete stop of melanophore differentiation at
the stage of S-melanoblasts (see 9 in Fig. 14). The always present
drosopterines become more visible in the skin, thus giving the fish
the "golden" coloration. This 9 mutation was introduced into the
hereditary melanoma-bearing hybrids by  Fig.15. Fish exhibiting crossing-conditioned hereditary melanoma in the compartment of the dorsal fin and X-ray-induced somatic mutation-conditioned melanoma in the compartment of the posterior part of the body (see Df and Pp in Fig. 13)  Fig.16. Littermates that segregate into "golden" (g/ g) animals (left) and tumor (g+ / g) animals (right). The 9 / 9 animals are protected from melanoma by a block of pigment-cell differentiation in the stage of the stem melanoblasts (see Fig. 14). Following treatment with promoters of cell differentiation (see Table 2) they develop melanoma similar to their heterozygous littermates introgressive breeding, i.e., by the replacement of the g+-bearing
chromosome by the g-bearing homolog. In animals heterozygous for
9 (g/ g+) the derepressed Tu oncogene still mediates melanoma formation
(Fig. 16, right). In the littermates that are homozygous for 9 (g/
g), however, no melanomas develop (Fig. 16, left): The block of
differentiation at the stage of the stem melanoblasts exerted by
the homozygous g-mutation protects the fish from melanoma formation
or from the activity of its own derepressed oncogene. To break this
protection mechanism we tested a large variety of mutagenic and
nonmutagenic agents. Most of these agents (see Table 2, column "Promotion")
promote almost simultaneously the differentiation of large amounts
of the noncompetent cells to the competent stage, which subsequently
become neoplastically transformed and give rise to melanomas. 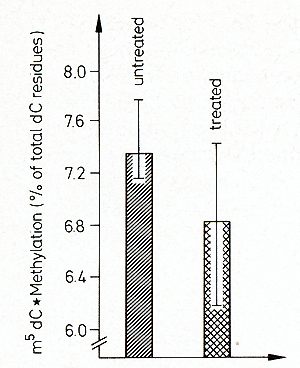 Fig.17. Decrease of m5dC methylation of DNA in Xiphophorus hybrids after treatment with 2.5 fig/liter per day 5-azacytidine. Fish were treated for 3 months, beginning at birth. HPLC analysis (filling, Supelco 4C-18-DB). Untreated, six animals; treated, five animals [63] These tumors are morphologically similar to the spontaneously
developing melanomas of the g/ g+ littermates, develop in the same
compartments of the body, and are, similar to all hereditary melanomas,
of multicellular origin. X-rays, N-methylN-nitrosourea (MNU), and
N-ethyl-Nnitrosourea (ENU), which are powerful mutagens or tumor
initiators, respectively, may also trigger melanoma by promotion
of cell differentiation. It appears, however, that in this case
neither of these agents acts as a mutagen, but as a differentiation-promoting
agent such as testosterone, TP A, and cyclamate, which are certainly
not mutagens. The assumption that any somatic mutation, including
that of a possible back mutation or suppressor mutation for the
9 mutation, is not involved in the promoting effect is compatible
with the multicellular origin of the melanoma and with the high
frequency of the respondents, which, in the case of 17 -methyltestosterone,
even reaches 100%. Optimization of the treatment in future experiments
will show whether the promoting effect of a certain agent is an
allor-llothing effect, as is conceivable on the basis of the concept
of this experiment. Since somatic mutations are probably not involved
in the trigger for melanoma development in this experiment, one
could speculate that the 9 mutation undergoes a general modification
that brings the function ofg close to the normal function of the
original g+. The promoting effect of 5-azacytidine could be interpreted
in this direction. This agent creates undermethylatioll in the DNA
and, as a consequence, presumably gene activation [9, 40, 41]. As
shown in Fig. 17, 5-azacytidine, which promotes differentiation,
also demethylates DNA of Xiphophorus. More data are required to
correlate the removal of the tumor-protecting effect of the g-mutation
by 5-azacytidine with DNA methylation. The crucial event leading
to neoplasia in this experiment is the promotion ofcell differentiation
and possibly a more general modification of a gene that is involved
in cell differentiation. The oncogene itself probably remained unchanged.
VII. Seventh Experiment: Tumors Induced by Elimination ofa Differentiation
Gene from the Germ Line and Impairment of That Gene in Somatic Cells
To study the effect of the differentiation gene (Diff), thousands
of melalloma-bearing BC segregants were produced according to the
crossing procedure shown in Fig. 5, and all showed a clear-cut I:
1 segregation into animals developing spontaneously benign (Fig.
5 E) or malignant melanomas (Fig. 5 F). BC hybrids that require
the carcinogenic trigger for the development of melanoma also show
a clear-cut Diffeffect (Fig. 11, see third experiment). Morphological,
histological, cytological, fine structural, biochemical, and molecular
studies showed that the majority of the cells of the benign melanoma
are well differentiated whereas those of the malignant melanoma
are poorly differentiated, and that differentiation of the transformed
cells is controlled by Diff [42, 43]. The most convincing data supporting
Diff-dependent control of pigment-cell differentiation in melanoma
come from transplantation experiments which have shown that pigmentcell
precursors present in the transplants taken from fish carrying the
deregulated Tu and lacking Diff (material of still tumorfree early
embryos of the malignant-mela-  Fig. 18. General cloverleaf structure of the tRNA
and positions of modified nucleosides. noma-developing genotype according to the fish shown in Fig. 5
F) become transformed and remain incompletely differentiated if
transplanted into embryos lacking Tu and Diff. The resulting animals
develop malignant melanoma. If, however, the pigment-cell precursors
of the same genotype are transplanted into Tu-lacking embryos that
contain the Diff gene, the cells of the developing melanoma become
terminally differentiated and regain their distance regulation:
These resulting animals develop extreme benign melanomas which regress
and eventually may become removed by macrophages (see [ I]). Further
studies on the Diff effect on melanoma in Xiphophorus were stimulated
by the experimental results of other laboratories that have focused
on the in volvement of nucleotide modifications of a certain tRNA
family in cell differentiation in eilbacteria, slime molds, and
cell cultures from different vertebrates [43-48]. These tRNAs include
tRNAAsn, tRNAAsp, tRNA His, and tRNA Tyr, which usually con tain
qileilosine (Q) instead of guanosine (G) in the first position of
the anticodon (position 34; see arrow in Fig. 18). The Q nucleoside
(7-(((4,5-cis-dihydroxy2-cyclopenten-l- yl) -amino) -methyl- 7 -deazagilanosine)
is unique in that its purine skeleton is modified to a 7 -deazastructure.
Eilbacteria synthesize the base qileuine de novo whereas vertebrates
are supplied with qileiline by nutrition or the intestinal flora.
Qileiline itself is inserted into the nucleotide chain of tRNA by
an exchange with guanine. This process is catalyzed by 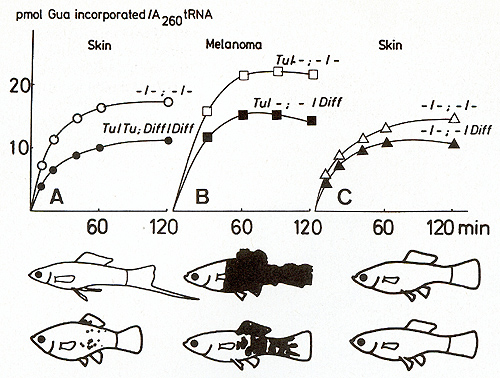 Fig.19A-C. Incorporation of [łH]guanine in position 34 oftRNA for Asp, Asn, His, and Tyr of Xiphophorus catalyzed by tRNA-guanine-transglycosylase (insertase) of E. coli. The graphs show the kinetics of the exchange of G34 of tRNA by [łH] guanine, a reaction used to evaluate the amount of (Q-)tRNA [50]. A-C according to the fish shown below the curves. These fish correspond to those shown in Fig, 5. High incorporation of łH] guanine in Diff-lacking animals corresponds to a low content of Q, whereas low incorporation of r H] guanine in Diff-containing animals corresponds to a high content of Q. A Skin of purebred Xiphophorus: white ring, X, helleri; black ring, X, maculatus. B Melanoma of BC segregants: white square , malignant; black square, benign. C Skin of nonmelanomatous BC segregants: white triangle, lacking Diff. Black triangle, containing Diff: Note that comparable Diff-containing animals always have a lower G content and a higher Q content than Diff-Iacking ones. Data from [43,44,51] tRNA-guanine-transglycosylases. The more the cells are differentiated,
the more replacement of G by Q is observed in position 34. The method
for estimating the G: Q ratio in a given population of the tRNA
family consisted of following the replacement of guanine in position
34 by a łH-labeled guanine exerted by a guanine-transglycosylase
(insertase) of Escherichia coli [50,51]. The results obtained in
Xiphophorus by measurement of [łH] guanine incorporation into the
tRNAs of the Q family, differing in the ratio of G : Q in position
34, are summarized in Fig. 19. The graphs show the kinetics of the
exchange of G 34 of the tRNA family by [łH] guanine, which is the
reaction used to evaluate the amount of (Q-)-tRNA. The fish genotypes
and phenotypes are identical to those shown in Fig. 5. In accordance
with the findings of many investigators working with other differentiation
systems [52], [łH] guanine incorporation is high in tRNAs from malignant
melanomas that consist predominantly of poorly differentiated cells.
In contrast, the incorporation is lower if the tRNAs are derived
from benign melanomas that consist predominantly of well-differentiated
cells. Therefore, tRNAs of malignant melanomas have a higher amount
of Gin place of Q than those of the benign melanomas (Fig. 19 B).
To decide whether the distinct difference in G: Q ratios between
benign and malignant melanoma is Diff' dependent or represents an
epiphenomenon of benignancy and malignancy, the skin of nontumorous
littermates that segregate into animals carrying Diff and lacking
Diff like the tumorous fish in a 1: 1 ratio was used for analysis
(Fig. 19C). The Diff-lacking segregants always had higher amounts
of Q-lacking tRNA than the Diff-carrying animals. The skin of the
parent animals used  Fig.20A,B. Development of induced somatic mutation-conditioned malignant melanomas on a crossing-conditioned superficial hereditary melanoma. A Untreated fish; B Fish treated with X-rays. It is assumed that the induced melanomas are due to an impairment of the differentiation gene Diff in a pigment-cell precursor each for the initial crosses showed the same differences (Fig. 19 A)
: X. helleri, which lacks the Diff gene, has a high łH] guanine
incorporation (i.e., is G-rich) whereas X. maculatus, which contains
the Diff gene, has a lower [łH] guanine incorporation. From these
results we suggest that the difference of G: Q ratios between benign
and malignant melanoma are not epiphenomena of benignancy and malignancy,
but are closely related to the primary effect of the Diff gene.
The differences in the functional properties of Q-containing and
Q-lacking tRNAs require further elucidation. The (Q)tRNAs have been
suggested to prefer codons NAU to NAC, whereas the Q-lacking tRNAs
read NAC and NAU equally well [46]. This may be an important mechanism
in the regulation of translation. For eukaryotic tRNA Tyr it has
been shown that the Q-lacking species reads a terminator codon,
probably UAG. Therefore the Q-lacking and Q-containing tRNAs of
vertebrates might select mRNAs for translation by a regulatory mechanism
similar to that of termination transcription control (see discussions
in ref. [51]). If benignancy depends on the presence of a single
copy of Diff and malignancy on the lack of this copy then it should
be possible to induce easily somatic mutation conditioned malignant
melanoma in the area of germinal-conditioned benign melanoma. For
this purpose we treated superficial benign melanoma with MNU or
X-rays and observed the development of focal malignant melanoma
in the area of the benign melanoma (Fig. 20). This is not to say
that the benign melanoma changes to the malignant state. In contrast,
the somatic mutation-conditioned malignant melanoma develops independently
from the already present germ-line-conditioned benign melanoma.
The induced malignant melanoma, however, competes with the hereditary
benign melanoma for pigmentcell precursors. As a consequence of
this competition the focal malignant melanomas are surrounded by
a halo-like zone that is sparsely polulated by the cells of the
benign melanoma. This experiment shows once more that the genes
that carcinogens act upon if they trigger neoplasia are not necessarily
the oncogenes themselves, but their regulatory genes in the broadest
sense. 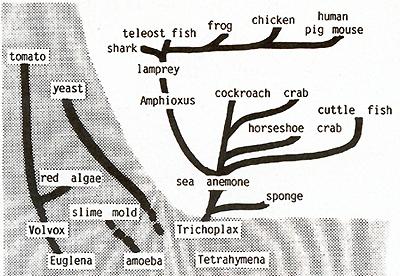 Fig.21. Distribution of the c-src oncogene in
living organisms. All organisms listed have been tested.
In our Xiphophorus model we have not found any genetic change of the Mendelian inherited oncogene Tu that might lead to neoplasia, although one would expect this to be possible based on the molecular findings of other laboratories that mutation, rearrangement, amplification, and demethylation can convert a silent oncogene to the transforming state. In contrast, our carcinogenesis studies show that the most important process involved in neoplasia in these animals is loss, impairment, or any other dysfunction of the regulatory gene system of the Tu oncogene. About 20 regulatory genes controlling the oncogene Tu at the gene level have been identified genetically and phenotypically. These genes comprise tissue-specific and compartment-specific (eye, mouth, etc.) regulatory genes (first to fifth experiments). Once the system of these regulatory genes controlling Tu is impaired, a chain of events is begun that can lead straight to neoplasia, but can also be interrupted by a genetic block of cell differentiation which protects the animal from the transforming activity of the oncogene. This protection mechanism can, however, easily be broken by promotion of cell differentiation (sixth experiment). If, finally, the cells are transformed, tumor growth can be stopped by terminal differentiation of the tumor cells exerted by the differentiation gene. Loss or impairment of the differentiation gene, then, leads definitely to neoplasia (seventh experiment). Our carcinogenesis experiments indicate that it is the regulatory genes (in the broadest sense ) and not the oncogene itself that the carcinogens commonly act on, when they trigger neoplasia in Xiphophorus. The inheritance and phenotypic expression of the genetically defined Tu oncogene parallels completely the expression of the molecularly defined c-src oncogene. Regardless of any future findings that might bring Tu substantially in relation to c-src and/ or any other c-onc, both Tu and c-src act, are regulated, and are inherited as if they were the same chromosomal gene of the natural gene pool of the Xiphophorus fish. c-src has been found functioning in all taxonomic groups of multicellular animals ranging from mammals down to the sponges (Fig.21) [16, 53], and intensive efforts are being made in many laboratories to determine whether a cellular oncogene, such as c-src, is capable of mediating neoplastic transformation like its viral counterpart [3, 54-59]. If this should be proven we suggest that all individuals of all metazoa are endowed with the capacity to develop neoplasia. Support for this idea comes from the fact that neoplasia is distributed -although sporadically -in all groups of multicellular animals [60-62]. Ubiquity of the oncogene in metazoa including humans on the one hand and the infrequent occurrence ofneoplasia in all these organisms on the other hand raises the question of the mechanisms that protect the majority of the individuals of all metazoans from the action of their own oncogenes. The Xiphophorus model provides an opportunity to contribute to the study of this problem.
Thanks are due to Kristine Krüger for valuable help in the preparation of this paper.
1. Anders F (1983) In: Neth R, Gallo RC, Greaves MF, Moore MAS, Winkler K (eds) Modern trends in human leukemia V. Springer, Berlin New York, pp 186-206 (Haematology and Blood Transfusion, vo128) 2. Reddy EP, Reynolds RK, Santos E, Barbacid M (1982) Nature 300: 149-152 3. Tabin CJ, Bradley SM, Bargmann CI, Weinberg RA, Papageorge AG, Scolnick EM, Dhar R, Lowy DR, Chang EH (1982) Nature 300:143-149 4. Taparowsky E, Suard Y, Fasano 0, Shimizu K, Goldfarb M, Wigler M (1982) Nature 300: 762-764 5. Payne GS, Bishop JM, Varmus HE (1982) Nature 295:209-213 6. Dalla Favera R, Wong-Staal F, Gallo R (1982) Nature 299:61-63 7. Schwab M, Alitalo K, Varmus HE, Bishop JM, George D (1983) Nature 303:497-501 8. Schimke RT (1984) Cancer Res 44: 1735-1742 9. Riggs AD, Jones PA (1983) Adv Cancer Res 40: 1-30 10. Pimentel E (1985) Cancer Genet Cytogenet 14:347-368 II. Kallman KD (1975) Handb Genet 4:81-132 12. Anders F (1967) Experientia 23: 1-10 13. Anders A, Anders F, Klinke K (1973) In: Schröder JH (ed) Genetics and mutagenesis offish. Springer, Berlin New York, pp 33-63 14. Anders F, Anders A, Vielkind U (1974) Int Cancer Congr I1th Florence 3:305 (Abstr) 15. Anders A, Anders F (1978) Biochim Biophys Acta 516:61-95 16. Anders F, Schartl M, Barnekow A, Anders A (1984) Adv Cancer Res 42: 191-275 17. Collet MS, Erikson RL (1978) Proc Natl Acad Sci USA 75: 2021- 2024 18. Barnekow A, Schartl M, Anders F, Bauer H (1982) Cancer Res 42: 2429-2433 19. Schartl M, Barnekow A, Bauer H, Anders F (1982) Cancer Res 42:4222-4227 20. Anders F, Schartl M, Bamekow A (1984) Natl Cancer Inst Monogr 65:97-109 21. Schartl M, Bamekow A (1984) Developmen tal Biology 105:415-422 22. Heil M ( 1984) Thesis, University of Giessen 23. Kollinger G, Schwab M, Anders F (1979) J Cancer Res Clin Oncol 95: 239-246 24. Berthiaume L (1984) Virology 135: 10-19 25. Vielkind U, Schlage W, Anders F (1977) Z Krebsforsch 90: 285-299 26. Ahuja MR, Schwab M, Anders F (1975) Ex perientia 31: 296-297 27. Mäueler W (1984) Thesis, University of Giessen 28. EI-Zawahri MM, Hamdoon NT, Anders F (to be published) Cancer Genet Cytogenet 29. Hamdoon NT (1984) Thesis, University of Giessen 30. Ahuja MR, Schwab M, Anders F (1980) J Hered 71: 403-407 31. Morizot DC, Sicliano MJ (1982) Genetics 102:539-566 32. Anders F, Schwab M, Scholl E (1981) In: Stich HF, San RHC (eds) Short-term tests for chemical carcinogens. Springer, Berlin New York, pp 399-407 33. Anders F, Schmidt CR, Herbert A, Anders A (1983) In: Prüfung von Chemikalien auf Kanzerogenität, Mutagenität und Teratogenität. Ges. f. Strahlen- und Umweltforschung, München, pp 253-274 34. Vielkind J, Haas-Andela H, Vielkind U, An ders F (1982) Mol Gen Genet 185: 379-389 35. Krontiris TG, Cooper GM (1981) Proc Natl Acad Sci USA 78: 1181-1184 36. Perucho M, Goldfarb M, Shimizu K, Lama C, Fogh J, Wigler M (1981) Cel127:467-476 37. Der CJ, Krontiris TG, Cooper GM (1982) Proc Natl Acad Sci USA 79:3637-3640 38. Anders F, Diehl H, Schwab M, Anders A (1979) In: Klaus SN (ed) Pigment cell; vol 4, Karger, Basel, pp 142-149 39. Anders F, Diehl H, Scholl E (1980) Linnean Soc Symp Ser 9:211-224 40. Constantinides PG, Taylor SM, Jones PA ( 1978) Dev Biology 66: 57- 71 41. Jones PA, Taylor SM (1981) Nucleic Acid Res 3:2933-2947 42. Vielkind U (1976) J Exp Zoo1196: 197-204 43. Anders A, Dess G, Nishimura S, Kersten H (to be published) In: Bagnara JT, Klaus S, Paul E, Schartl M (eds) Pigment Ce111985 44. Kersten H (1982) In: Jaenicke L (ed) Bio chemistry of differentiation and morphogen esis. Springer, Berlin New York, pp 116-119 45. Kersten H (1983) in: Nass G (ed) Recent re sults in cancer research: modified nuc leosides and cancer. Springer, Berlin New York, pp 255-263 46. Nishimura S (1983) Prog Nucleic Acid Res Mol BioI 28:49-74 47. Katze JR, Beck WT, Cheng CS, McC1oskey JA (1983) In: Nass G (ed) Recent results in cancer research: modified nucleosides and cancer. Springer, Berlin New York, pp 146 -159 48. Katze JR, Gündüz U, Smith DL, Cheng CS, McCloskey JA (1984) Biochemistry 23: 1171 -1176 49. Sprinzl M, Gauss DH ( 1982) Nucleic Acids Res 10:rl-r55 50. Okada N, Nishimura S (1979) J BioI Chem 254:3062-3066 51. Kersten H, Schachner E, Dess G, Anders A, Nishimura S, Shindo-Okada N ( 1983) In: Curtis HC, Pfleiderer W, Wachter H (eds) Biochemical and pteridines. Walter pp 367-382 52. Nass G (1983) Recent results in cancer re search. Modified nucleosides and cancer. Springer, Berlin New York 53. Barnekow A, Schartl M (1984) Mol Cell Biol 4: 1179-1181 54. Takeya T, Hanafusa H (1982) J Virol 44: 12-18 55. de Klein A, van Kessel AG, Grosveld G. Bartrarn CR. Hagemeijer A, Bootsma D, Spurr NK, Heisterkamp N. Groffen J, Stephenson JR ( 1982) Nature 300: 765- 767 56. Duesberg P (1983) Nature 304:219-225 57. Bishop JM (1982) Sci Am 246:68-78 58. Weinberg RA (1983) Sci Am 249: 102-116 59. Hunter T (1984) Sci Am 251 :60-69 60. Kraybill HF, Dawe C.J, Harshbarger .JC. Tar diffRG (1977) Ann NY Acad Sci 298 61. Kaiser HE (1981) Neoplasms -comparative pathology of growth in animals, plants and man. Williams and Wilkins, Baltimore 62. Dawe Cl, Harshbarger .JC. Kondo S, Sugimura T, Takayama S (1981) Phyletic approaches to cancer. Japan Scientific Societies Press, Tokyo 63. Schmiedel A (1984) Thesis. University of Giessen |