|
* This contribution appears also in the 33rd Mosbach Colloquium: "The Biochemistry and Differentiation of Morphogenesis"' also published by Springer Xiphophorus, including swordtails and platyfish, is a genus of small viviparous freshwater fish from Central America that serves increasingly as a laboratory animal [I]. For 25 years we have used Xlphophorus for studies on neoplasia, which can easily be induced in hybrids between descendants of different provenance [2]. Although neoplasia of these animals is rather well understood in terms of formal genetics, the molecular basis of this phenomenon was extremely resistant to any elucidation. Recentlya promising approach to the study of neoplasia in Xiphophorus at the molecular level has been undertaken in a cooperative work of the laboratories of H. Bauer (Institut für Virologie, Giessen), w. and H. Kersten (Institut für Physiologische Chemie, Erlangen), S. Nishimura (National Cancer Center Research Institute, Tokyo ), and our laboratories. The present review will trace some steps of our studies that led to the detection of a cellular oncogene and a prominent regulatory gene. A. The Taxonomic Groups of Xiphophorus The genus Xiphophorus lives in genetically isolated populations in brooks, rivers, lakes, ponds, and pools, and has evolved into innumerable genotypically and phenotypically distinguishable groups [ I]. Based on certain morphological and ecological characters, 17 of these groups have been listed as species [3]. All individuals of this genus, however, can be hybridized in the laboratory without difficulty, and all hybrids are fertile. This, together with the findings on the conformity of genome organization [4], the low degree of enzyme polymorphism [5, 6], and the normal chromosome pairing during meiosis in the hybrids [7], led to the conclusion that the taxonomic differences between these groups of Xiphophorus are not at the species level, but at the level of elementary local populations as well as ecological and geographical races.
Tens of thousands of individuals from different wild populations of Xiphophorus have been collected by several investigators (see [2]), but no tumors were detected. In the progeny of the wild populations, which in the case of X. helleri from Rio Lancetilla and X. maculatus from Rio Jamapa have been bred in the laboratory since 1939 (about 80 and 120 generations, respectively), no tumors occurred. About 10,000 specimens of purebred descendants of the wild populations have been treated with powerful carcinogens such as benzo(a)pyrene, N-methyl-N-nitrosourea (MNU), and X-rays, but none developed neoplasia. These animals are highly insusceptible to neoplasia (Table I, first part). Table 1. Neoplasia in Xiphophorus 1 year
after treatment with MNU (10-ł M; four times for 1 h at 2-week intervals)

In contrast to the animals from purebred wild populations, animals from laboratory hybrid populations derived from crosses between the purebred descendants of wild populations may be susceptible to neoplasia. Following treatment with carcinogens, depending on the wild populations used for hybridization, about 1%-4% of the Fl-hybrids develop neoplasia. Tumor incidence increases in the second hybrid generation (F 2) up to about 10% and remains stable in the succeeding generations, which have been tested up to F24 (Table I, second part).
As compiled from the results of several investigators in our laboratories [8] 805 of 10,195 (8% ) hybrids which survived treatment with MNU and X-rays developed a large variety of different neoplasms. Most of the neoplasms were classified as neurogenic and mesenchymal, with melanoma, neuroblastoma, and fibrosarcoma being the predominant types. Epithelial neoplasms were less frequent but comprised the largest diversity (Table 2). E. Assignment of Cancer Susceptibility to Chromosomes To study what may be the crucial differences between the fish that were insusceptible and those that were susceptible to cancer, we attempted to assign the carcinogen-triggered neoplasms to chromosomes. For this study 65 defined genotypes of x. maculatus, X. xiphidium, X. variatus, X. cortezi, X. helleri, and their hybrids were employed [9, 10]. The genotypes exhibit, or lack, specific colour patterns or enzyme markers which are due to the expression of specific genes, of which each is located on a different chromosome. We used mainly backcrosses, which were selectively bred for a specific phenotypic marker, and thereby for a specific chromosome. Such backcrosses segregate into 50% animals carrying the marker chromosome, and 50% lacking this chromosome. Neoplasia could be assigned specifically to many different chromosomes. In the example shown in the scheme of Fig. I almost exclusively the backcross (BC) segregants exhibiting the stripes inherited from x. maculatus were susceptible. They developed melanoma, neuroblastoma, ephithelioma, and fibrosarcoma. Some of these hybrids developed several tumors of different types. This is not to say that all fish exhibiting the stripes develop neoplasia; but almost all neoplasms develop in those animals that belong to the group of Table 2. Neoplasms induced in F2-
F24 and BCn generations (MNU: 6608 survivors; X-rays: 3587 survivors;
total: 10, 195) 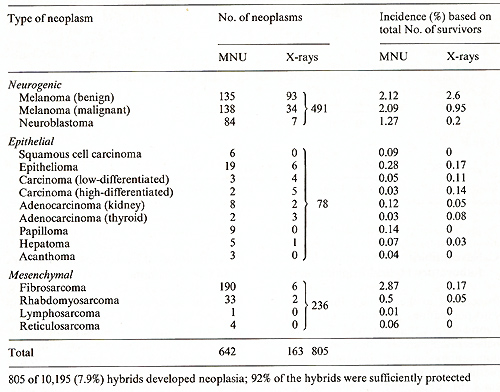 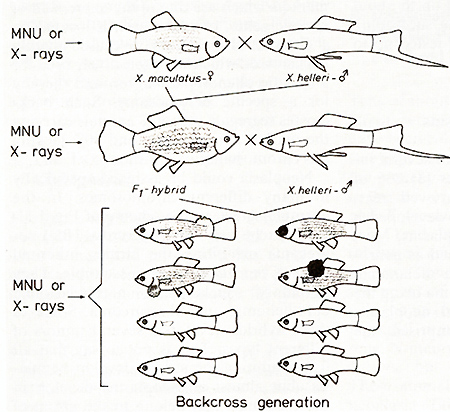 Fig. I. Cyossing scheme showing the assignment
of carcinogentriggered neoplasms to backcross segregants exhibitingstripes BC segregants exhibiting stripes. Thus, the susceptibility to develop different kinds of neoplasms apparently depends on the marker chr0mosome which is responsible for the stripes. There are other examples in which susceptibility to neurogenic and epithelial neoplasia could be assigned to a certain chromosome while susceptibility to neoplasms of mesenchymal origin depends on a different chromosome [10]. Other examples have shown that susceptibility to develop melanoma may depend on aY chromosome, an X-chromosome, or an autosome. For all backcross hybrids tested so far, susceptibility to develop neoplasia is apparently chromosome specific.
Information about the genes underlying susceptibility to carcinogen-triggered
neoplasia comes from the analysis of the hereditary trait of certain
"spontaneously" developing melanomas, pterinophoromas, neuroblastomas,
thyroid carcinomas, kidney adenocarcinomas, and reticulosarcomas
in hybrids. These neoplasms are very rare compared with the carcinogentriggered
tumors and therefore have been considered as a curiosity by many
cancer researchers. They can, however, be produced at the will of
the experimenter and have therefore been studied by many investigators
[I]. They have contributed many important facts for developing ideas
about how other animals, including humans, might inherit cancer
[11, 12]. In the following chapters we shall mainly deal with the
genes involved in melanoma development because their phenotypic
effect, due to the enlarged shape and heavy pigmentation of the
transformed cells, can easily be observed without killing the fish.
Even a single transformed cell can be distinguished from a regular
pigment cell. CIosses of a spotted x. maculatus (platyfish) with
a nonspotted x. helleri (swordtail) result in F 1-hybrids that develop
benign melanoma instead of spots (Figs. 2, 3). Backcrosses of the
F 1-hybrids using the swordtail as the recurrent parent result in
offspring (BC1), 50% of which exhibit neither spots nor melanoma
while 25% de velop benign melanoma (like the F 1), and 25% develop
malginant melanoma. Further backcrosses of the fish (not shown in
Fig. 3) carrying benign melanoma with the swordtail result in a
BC2 that exhibits the same segregation as the BC1. The same applies
for further backcrosses. Backcrossing of the fish carrying malignant
melanoma with the swordtail results in a BC2 in which 50% of the
animals do not develop melanoma, while the remaining 50% develop
malignant melanoma. In contrast, backcrossing of the melanoma-bearing
hybrids using the platyfish as the recurrent parent results in a
gradual suppression and finally disappearance of neoplasia in the
succeeding generations. These results, with the inclusion of cytogenetic
findings, were interpreted as follows [2]: The spots and their corresponding
genes are a specific accessory of the platyfish. The melanomas of
the hybrids are closely related to the spots which actually are
extreme benign melanomas. On the other hand the swordtail lacks
both the corresponding spots and genes (Fig. 3). The genetic information
for neoplastic transformation of pigment cells in encoded in a "tumor
gene" (Tu) which is inherited by the platyfish. About 70 crossovers,
deletions, duplications, and translocations show that Tu is located
at the end of the X-chromosome and is under control of linked and
nonlinked regulatory genes [ 13]. In the platyfish used in this
experiment a "major" pigment cell-specific regulatory gene (R) linked
to Tu as well as two "minor" regulatory genes, which compartment-specifically
suppress melanoma formation in the dorsal fin (RDf and the posterior
part of the body (Rpp), are mutated to R', RDf', and Rpp', respectively,
and can no longer suppress Tu. Evidence for this comes from the
appearance of some transformed pigment cells in the dorsal fin and
in the skin of the posterior part of the body. The regulatory gene
that actually suppresses tumor formation in the platyfish used in
this experiment is the homozygous nonlinked "differentiation gene"
(Diff which can easily be detected by the esterase marker (Est-])
closely linked to Diff[6, 14, 15]. Further regulatory genes also
present in the system are not taken into consideration.  Fig.2A-D. Spots and melanomas in Xiphophorus. A X. maculatus from Rio Jamapa (Mexico); "spotted dorsal fin" (mutation R Dl) and "spotted posterior part" of the body (mutation R Pp`). B F 1-hybrid between X. maculatus A and X. helleri from Rio Lacetilla (Honduras) exhibiting benign melanoma instead of spots. C BC-hybrid B with X. helleri as the recurrent parent exhibiting malignant melanoma instead of spots. D Albino BC-hybrid exhibing malignant amelanotic melanoma. A-C correspond to the respective schematic drawings shown in Fig. 3 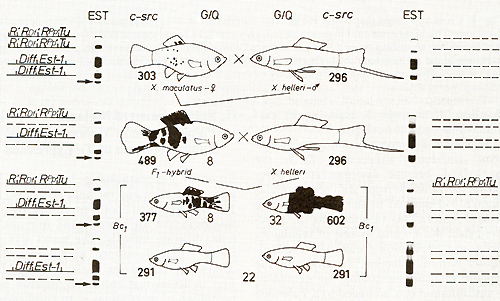 Fig.3. Crossing scheme which displays the genetic conditions for the "spontaneous" development of spots (Fig. 2 A), benign melanoma (Fig. 2 B), and malignant melanoma (Fig. 2 C). -chromosomes of X. maculatus; chromosomes of X. helleri,. Tu, tumor gene; R Pp and R Dl`f impaired regulatory genes controlling Tu in the compartments of the posterior part of the body (Pp) and the dorsal fin (Df); R'`f impaired regulatory gene specific to pigment cells but nonspecific to the compartments; Diff, regulatory gene controlling differentiation of neoplastically transformed cells; Est-l`f locus for esterase I of X. maculatus. EST. esterases (polyacrylamide gel electrophoresis from homogenates of the eye ); note linkage of Diff and Est-l (see arrow.s). c-src, pp60c-src kinase activity ( cpm/mg protein; 53K; see Fig. II); note basic and excessive activity`f and correlation between c-src expression and Tu expression. G/Q`f łH-guanine incorporation in position 34 (anticodon) of tRNAs for Tyr, Asn, Asp, and His (pmol/ A26o ; see Fig. 15); note low incorporation in tumors of Diff animals indicating high Q content, and high incorporation in the melanoma of Diff-lacking animals, indicating low Q content (combined from data of[2`f 6`f 13, 14,30, 31,34,40,46]). For details see text
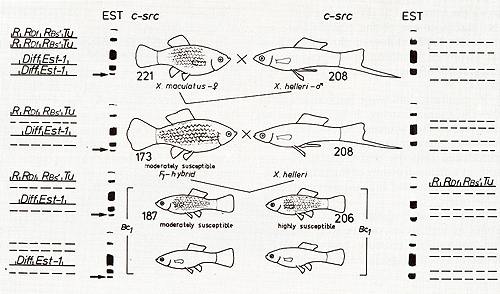 Fig. 4. Crossing scheme displaying the genetic conditions for susceptibility to carcinogen-dependent neoplasia. The highly susceptible genotype is highly sensitive to the carcinogenic (mutagenic) trigger. Abbreviations according to Fig.3 Bs`, impaired the regulatory gene controlling Tu in the compartment of the entire side of the body (Bs) (combined from data of[6, 16,31,34]). For details see text deletion, of a single R gene by the carcinogen in a somatic cell [16]. In conclusion, the regulating gene systems confidently suppressing Tu in the nonhybrids become disorganized if chromosomes derived from different populations are combined in the hybrids by the experimenter. This implies that, in purebred fish, the Tu and its sets of regulatory genes are population-specifically coadapted by natural selection. In any case the genetic information coding for neoplastic transformation can be traced to a Tu which is present in the different cell types and is normally under control of population-specific and cell-type-specific polygenic systems of linked and nonlinked regulatory genes, which suppress the development of the various types of potential neoplasms (Fig. 5). According to the formal assignment of different neoplasms to a particular chromosome, the particular Tu that codes for neoplastic transformation of pigment cells may also code for transformation of cells of mesenchymal and epithelial origin (see Figs. 1, 5).
To study the relationship between melanoma formation and melanin
synthesis [ 17, 18] we separated both processes by introgression
of an albino gene into melanoma-bearing hybrids. The result was
albinos which developed melanomas that completely lacked the melanin
(Fig. 2 D). This indicates that enhanced melanin pigmentation of
melanomas is an epiphenomenon of neoplastic transformation of pigment
cells exerted by Tu. 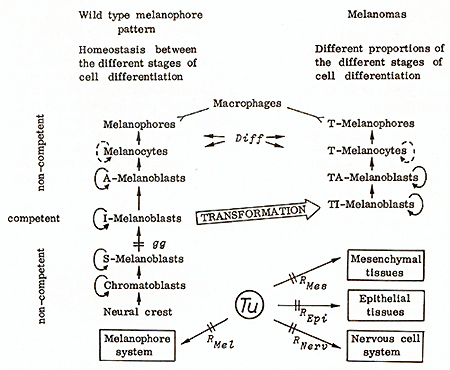
The precursors of the melanin-producing pigment cells of Xiphophorus, like those of other vertebrates, originate from the neural crest, and migrate to their final destination (see Fig. 5). They divide and undergo differentiation through the stages of chromatoblasts, stem(S)-melanoblasts, intermedia te(I)-melano blasts, advanced( A )- melanoblasts, melanocytes, and, finally, differentiate to melanophores, which are incapable of dividing. At a certain age the melanophores are removed by macrophages. Supply comes from S-melanoblasts [2, 19,20]. A-melanoblasts, melanocytes, and melanophores have never been observed to undergo neoplastic transformation. Thus, these cells appear to be noncompetent for the Tu activity. On the other hand, in genotypes carrying a mutation that arrests differentiation at the stage of S-melanoblasts, melanomas cannot develop until exogenous promoters push differentiation of S-melanoblasts to the stage of I-melanoblasts [21]. These studies show that also the neural crest cells, chromatoblasts, and S-melanoblasts are noncompetent. We therefore conclude that the only stage of differentiation in which the pigment cells are competent for the transforming activity of Tu is the stage of I-melanoblasts. These cells become transformed to TI-melanoblasts. We assume that the principle of competence of a cell to the transforming activity of Tu applies also for other kinds of neoplasms. It has not been possible to show so far whether Tu is still active in the transformed cells (T cells) for the maintenance of the neoplastic state. In any case the TImelanoblasts continue to differentiate to TA-melanoblasts, T -melanocytes and, finally, to T -melanophores that are incapable of dividing. This process corresponds to differentiation of the nontransformed pigment cells. It is not under the control of Tu but under the control of the differentiation gene Diff This, furthermore, implies that Tu does not specify the degree of malignancy of the melanoma (see later).
Information about the genuine effect of Tu come.s from a balanced
laboratory stock carrying a leth~l Tu translocation that origInated
according to Fig. 6. Tu from an X-chromosome of x. maculatus becomes
translocated to an autosome of x. helleri and, in its new position,
is not under control of its former linked regulatory genes (R, RDJ,
etc.). The progeny of this stock segregates into 50% carrying the
nonlinked D iff which survive, while the corresponding 50% lacking
Diff is lethal. As a consequence of the Mendelian inheritance of
the Tu translocation through the germ line, and the lack of Diff
Tu becomes active in the developing progeny as soon as the pigment
cell precursors differentiate to the competent I-melanoblasts. This
process starts in the 5-day-old embryos. Some time later some single
dividing T -melanocytes appear at the peduncle of the tail fin of
the embryo (Fig.7). Neoplastic transformation continues in all areas
of the developing embryo, where a pigment cell precursor becomes
competent, thus building the lethal "whole body melanoma". This
reflects the genuine effect of the completely derepressed Tu on
the pigment cell system [13]. It is suggestive to assume that Tu
might exert an essential function in the early embryo which is related
to the neural crest a~d its. derivatives. In normal embryogenesIs
thIS functIon becomes switched off or choked by the regulatory genes
prior to the 5th day of embryonic life. If, however, the regulatory
genes (i.e., the entire switch) are lacking, Tu continues to exert
its early embryo-specific function, which as a process of misguided
cellular development transforms the competent cells to the neoplastic
state. 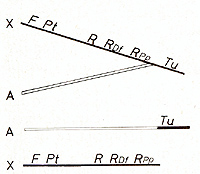
In the sex chromosomes of the platyfish 30 deletions involving
Tu have been characterized genetically, and some of the major deletions
involving both the Tu and its linked regulatory genes, additionally
to the genetic results, were cytologically observed [ 13, 22]. All
deletions are nonlethal. Even the loss of a Tu-containing segment
of the X-chromosome (one Giemsa band), in the homozygous condition
in the female or in the hemizygous state in the male, apparently
has no detectable effect on viability. This, together with the fact
that the swordtail used in our crossing experiments populatlonspeclfically
lacks the Tu (see Figs. 3, 4), led us to the conclusion that the
Tu so far considered is not essential but is an accessory to the
fish [2]. This is not to say that the Tu has no normal function.
One could, for instance, assume that additional copies of Tu that
are indispensable to the fish are present in the autosomes and may
compensate for the loss of the sex chromosomelinked Tu loci according
to a gene dosage compensation mechanism which warrants normal functions.
Support for the assumption of multiple copies of Tu per haplOId
genome comes from the following experment. Platyfish, carrying the
deletion of the Giemsa band that involves the accessory Tu, were
crossed according to the procedure outlined in Figs. 3, 4, with
the swordtail populationspecifically lacking the accessory Tu. No
tumors developed in the hybrids. Following treatment of the backcross
hybrids with MNU, however, melanomas developed which were specifically
localized at the upper part of the tail fin. These neoplasms could
be assigned to an autosome. Thus it appears that the platyfish,
besides the easily detectable accessory Tu copies contains additional
ones that require more intricate experiments for their detection.
Our experience that all individuals of all groups of Xiphophorus
can contribute to susceptibility to neoplasia in the hybrids suggests
that all individuals contain Tu copies that are indispensable for
the fish, and may contain accessory Tu copies. Up to ten accessory
copies of the repressed Tu could be introduced into a laboratory
stock by crossings. N o effect on viability could be observed. 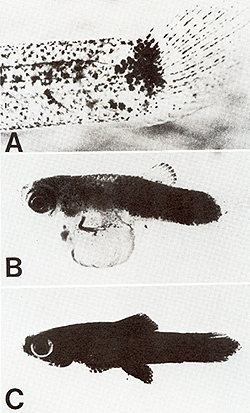
Both the X-chromosome of x. maculatus containing Tu (the X-chromosome
according to Fig. 3) and the X-chromosome of x. maculatus, having
lost the Giemsa band carrying Tu, were introduced into the ge nome
of X. helleri lacking the regulatory genes for the accessory Tu.
XTu X Del X XTu y matings were accomplished. The segegating offspring
had none, one, or two, respectively, accessory Tu copies and showed
a clear-cut gene dosage effect (Fig. 8). If, however, the experiment
was modified by using animals as recipients, having retained the
nonlinked regulatory genes, we observed a clear-cut gene dosage
compensation (Fig.9). Dosage effect and dosage compensation of the
accessory oncogene Tu, therefore, depends on the absence or presence,
of the nonlinked regulatory genes. Oncogene dosage effect and oncogene
dosage compensation has been observed in many experiments of this
kind [23].  Fig. 8A-C. Gene-dosage effect of the (incompletely) derepressed tumor gene (Tu). A No Tu (not a single transformed pigment cell). B One dosage of Tu (melanoma formation). C Double dosage of Tu (double effect in melanoma formation). Compare with Fig. 9  Fig.9A-C. Gene dosage compensation of the repressed tumor gene (Tu). A No Tu (not a single transformed pigment cell). B One dosage of Tu (spots consisting of transformed pigment cells; see dorsal fin). C Double dosage of Tu (effect shows no difference to that of one dosage). Compare with Fig. 8 DNA from laboratory platyfish carrying several accessory copies
of Tu (derepressed as well as repressed) was injected into the neural
crest region of early embryos of the swordtail which lacked both
the accessory Tu copies and the regulatory genes (Fig. 10). The
injected DNA may maintain its high molecular weight for about 2
hand thereafter becomes degraded to pieces which are too small to
contain genetic information [24]. Following the successful uptake
of Tu by an S-melanoblast of the embryo, this cell may later become
competent to the Tu activity by differentiation to an I-melanoblast,
which eventually may become neoplastically transformed to a TImelanoblast.
Additional proliferation of T cells amplifies the original transforming
effect of Tu, and the result becomes visible as the transformed
cells differentiate to the easily detectable colonies of T -melanocytes
and T -melanophores [25]. Depending on the type of the Tu donor
DNA ( cotransfer of intact or damaged regulatory genes) the percentage
of recipients showing T -melanocytes and T -melanophores ranged
from 0.4% to almost 8% (total number of survivors tested in these
experiments: 1390). Since the number of the target cells (pigment
cell precursors) at the time of DNA injection has been estimated
to be about 1000, the frequency of the transforming event on the
cellular basis is in the range of 10high - 55 [25]. This is the
same order of transfection frequencies reported for cell culture
systems [25-27]. 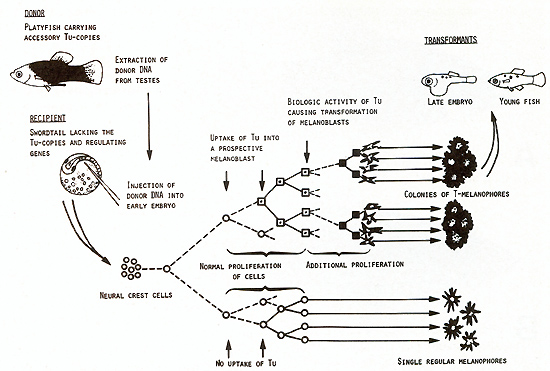
Several virological observations came to our knowledge which could
be of interest for a molecular interpretation of the Tu gene: In
chicken it was found that the oncogene v-src from Rous sarcoma virus
(RSV) has a counterpart, c-src, in the noninfected cells [28]. C
-src or at least a similar gene was also found in mammals including
mouse, calf, and humans [29]; and commercial DNA derived from salmon
[29] obviously contains the same gene. There is, however, no convincing
evidence to relate the cellular src or its gene product, a 60,000-dalton
phosphoprotein with kinase activity (pp60c-src), to neoplasia that
depends on conditions other than virus in fections (see discussions
in [30, 31]). With this background we started the search for c-src
in the genome of Xiphophorus. C-src was detected in Xiphophorus
by molecular hybridization of a src-specific probe from cloned v-src
with DNA from fish [32]. To identify pp60c-src, brains of the fish
were labeled with 32P-orthophosphate, and brain extracts were immunoprecipitated
with antisera from RSV tumor bearing rabbis (TBR-serum) followed
by polyacrylamide gel electrophoresis. The 60K protein detected
in the gel has a tyrosinespecific kinase activity, and represents
the pp60c-src [31 ]. The kinase activity was measured ac cording
to Fig. 11 [33] and then determined (see legends of Figs. 3, 11)
in several tissues including skin, liver, spleen, testes, brain,
and melanoma. Brain and melanoma always had the highest kinase activity.
Genotype-specific differences in kinase activity showed an identical
trend in both brain and melanoma [31]. To compare c-src expression
in nontumorous and tumorous fish, kinase activity was mainly determined
in the brains of these fish. 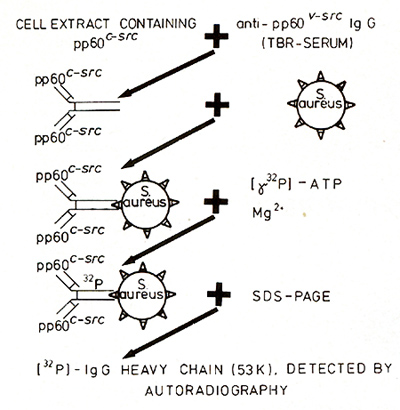
To study the possible relation between neoplasia and c-src expression we manipulated neoplasia in Xiphophorus according to the three genetic experiments outlined in Figs. 3,4 and 12 [34]. 1. In the experiment recorded in Fig. 3 the purebred X. maculatus carrying two repressed copies of the accessory Tu, as well as the purebred X. helleri and the BChybrids lacking the Tu, display the same activity of c-src kinase. This activity appears to be the basic expression of c-src. In contrast, the melanoma-bearing hybrids which contain the derepressed Tu show an increase of c-src activity, with the malignant melanoma bearing BC-hybrids displaying the highest activities. 2. In the experiment recorded in Fig.4 all purebred and hybrid animals, irrespective of the lack and the dosage of Tu but dependent upon the nontumorous state exerted either by several regulatory genes or by a linked R alone (see the highly susceptible genotype ), display a uniform c-src activity which seems to represent the basic c-src expression, as in the purebred animals and Tu lacking hybrids in Fig. 3. 3. In littermates (Fig. 12) which are genetically identical except for the lack of the accessory Tu and the presence of one or two partially de repressed accessory Tu copies, c-src displays a kinase activity that increases stepwise in parallel to the lack and the dosage of Tu, which, in their turn, determine whether the animals will develop no tumors, slowly growing tumors, or fast-growing tumors. Table 3 shows additional experiments of the same kind that yielded similar results. The main results of these experiments are that the nontumorous fish display a basic expression of c-src which in the tumorous fish may increase stepwise under two different conditions, namely (a) the stepwise derepression of an accessory Tu, and (b) the stepwise introduction of additional copies of a derepressed accessory Tu. Since the measurements were accomplished in the brains of the fish the increase of the activity of c-src is related di Table 3. pp60c-src associated kinase ativity
in brain extracts specified by cpm per milligram  rectly to the activity of Tu, and does not represent an epiphenomenon of melanoma formation, such as the elevated activity of many enzymes, [5,6, 14, 15] enhanced melanin synthesis [ 18], certain chromosome aberrations [35], etc. These findings suggest several possibilities for an interpretation of how Tu might be related to c-src: (a) Tu might be independent from c-src, and the correspondence between both Tu and c-src is due to linkage relationship. (b) c-src might represent a regulatory gene for Tu or vice versa. ( c) Tu might consist of different oncogenes that are responsible for different kinds of neoplasia and c-src is one of these genes. ( d) Tu might be identical to c-src, and this oncogene is capable ofcoding for a large variety of neoplasia. At present we cannot decide on a special interpretation. Additional data are required.  Fig. 12 A-C. Correlation between gene dosage effect of Tu (phenotype of the tumor) and gene dosage effect of c-src (pp60c-src kinase activity) in littermates containing A no accessory Tu, B one dosage of Tu, C double dosage of Tu. The genetic backgrounds of the fish are identical. A Tu is deleted in the germ line. B, C The pigment cell-specific R linked to Tu is impaired by germ line mutation [13, 23]. Kinase data from [34]
Table 4. Expression of pp60c-src kinase
in brain extracts of different fish species [31 ]
The presence of c-src in the genome of different animals such as chicken, salmon [29], and Xiphophorus led to the more systematic search for this oncogene in additional taxonomic groups of animals. Firstly, different groups of Xiphophorus and different fish genera more or less related to Xiphophorus were investigated. All fish tested (Table 4) show a pp60c-src kinase activity indicating that c-src must be present [31]. In addition, c-src was evidenced by its kinase activity in a large variety of metazoans other than fish, ranging from mammals to sponges, which, together with the results from other laboratories are listed in Table 5. C-src was not found in protozoa, algae, or higher plants [36]. The distribution of the cellular counterpart of the virl v-src brings about the idea that c-src might have evolved together with the multicellular organization of the animals, and that neoplasia might be a character that is closely related to this evolution. The gene Diff is one of the most prominent regulatory genes known in the melanoma system of Xiphophorus. As shown in Fig. 3, benignancy and malignancy in the hybridization-conditioned melanomas depend upon the presence or absence of the chromosome carrying Diff Biochemical markers for this chromosome, i.e., the esterase Est-J and the isozyme A of the glyceraldehyde-3-phosphate dehydrogenase [6] have confirmed that the Diff-carrying chromosome is derived from the platyfish, the source of accessory Tu. Table 5. c-src in eukaryotes (from [36])

The clear-cut Mendelian segregation of benignancy and malignancy has provided the opportunity to study the basic differences between the benign and the malignant state of the melanomas (Fig. 13, Table 6). The cytological, fine structural, biochemical, and biological data suggest that Diff promotes the differentiation of T cells: If Diff is lacking, the majority of the melanoma cells persist in the stage of the poorly differentiated, continuously dividing T Amelanoblasts and T -melanocytes, and only few cells differentiate to the final stage of the T -melanophores. If, however, the Diff is present, the majority of the melanoma cells become terminally differentiated to T -melanophores, whereas only a few cells remain in the stage of T A-melanoblasts and T -melanocytes. The T -melanophores at a certain age are removed by macrophages ([14, 18, 20, 38], see also Fig.5). This process is antagonistic to the permanent supply of melanoma cells from S-melanoblasts, and thus the melanoma is rendered benign [20, 21 ].
Transplantation experiments, including the composition of chimeras by fusion of parts of early embryos, have shown that pigment cell precursors present in the transplants taken from fish carrying Tu but lacking Diff (see Fig. 3) become incompletely differentiated and give rise to malignant melanoma if transplanted into embryos lacking Tu and Diff If, however, the pigment cell precursors of the same genotype were transplanted into Tu-lacking embryos that contain the Diff the cells of the developing melanoma become terminally differentiated and regain their distance regulation (Fig. 14). Thus the effect of Diffon the differentiation of the neoplastically transformed pigment cells can be traced to a dif fusible substance [39]. The nature of this substance is unknown. Table 6. The Gene Diffin Tu/ -Xiphophorus
[2,6, 14, 18,20,21,38,39,40,46] 
There is considerable evidence for the involvement of tRNA containing
modifications of the nucleotides in the process of cell differentiation
in normal and neoplastic tissues [40]. Many studies were focused
on a family of tRNAs including tRNAAsn tRNAAsp tRNA HIS and tRNA
Tyr which may contain queuosine (Q) instead of guanosine (G) in
the first position of the anticodon (position 34). Q is a hypermodified
G. The more the differentiation progresses, the more G is replaced
by Q in position 34 [40-46]. The method to estimate the G : Q ratio
in a given popu- 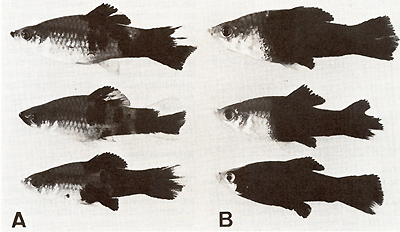 Fig. 13 A, B. Segregants of littermates A containing one dosage of the differentiation gene (Diff) and B lacking Diff Segregation of animals carrying benign and malignant melanoma is according to that of the backcross generation shown in the schematic drawings of Fig. 3  Fig. 14. Secondary chimera composed by transplantation of tissues containing precursor cells of malignant melanoma that originated from a young BC-hybrid containing Tu but lacking Diff (see malignant melanoma developing BC-segregant in Fig. 3), to a littermate lacking Tu but containing Diff (see the nontumorous BC-segregant at bottom left in Fig. 3). Note terminal differentiation and distance ( density) regulation of the transformed cells of the transplant according to the Diff genotype of the host (from [39]). For details see text lation of the tRNA family consists in following the replacement
of guanine in position 34 by a labeled guanine exerted by a guanine-transglycosylase
of E. coli. We have measured the incorporation of łH-guanine in
the tRNA for Asn, Asp, His, und Tyr in the malignant melanoma, the
benign melanoma, and the skin of melanoma-free littermates. In addition,
F 1hybrids carrying benign melanomas were studied ([40,46], Figs.
3, 15): łH-guanine incorporation is high if the tRNAs are prepared
from malignant melanomas (predominantly poorly differentiated cells).
In contrast, the incorporation is low if the tRNAs are derived from
benign melanomas (predominantly well-differentiated cells). The
tRNA family of the malignant melanomas, therefore, is G-rich, whereas
the tRNA family of the benign melanomas is Q-rich. łH-guanine incorporation
ill the skin of nontumorous littermates is intermediate between
those of the malignant and the benign melanomas. Since the nontumorous
fish (like the tumorous ones) consist of individuals lacking and
containing Diff in a I: I ratio, it is suggestive to assume that
the intermediate data represent a mean value of łH-guanine incorporation
in the Diff containing group and in the Diff-lacking group. If this
is correct the different a: Q ratios in the different melanomas
are no epiphenomena of malignancy and benignancy, but are very closely
related to the primary effect of Diff that in tumorous fish converts
the malignant to the benign state. 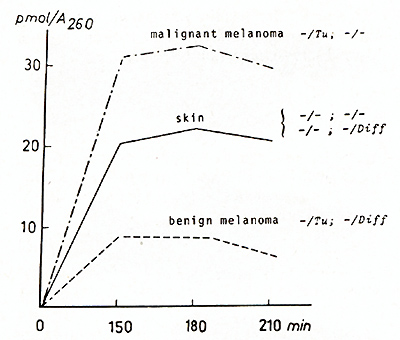 Fig. 15. Incorporation of łH-guanine in position 34 (anticodon) of tRNAs for Tyr, Asn, Asp, and His catalyzed by tRNA-guanine-transglycosylase (insertase) of E. coli. The melanomas (both malignant and benign) were derived from littermates of BC-hybrids according to Fig. 13 or Fig. 3, respectively. The skin was derived from both Diffcontaining and Difflacking nontumorous BC-segregants according to Fig. 3. Compare with G /Q data shown in Fig. 3 (from [40, 46]). F or details see text
We have studied neoplasia of Xiphophorus at different levels of the biological organization including species, races, populations, generations, littermates, individuals, tissues, cells, genomes, chromosomes, and genes. In doing so we could trace neoplastic transformation to the activity of one or several copies of the oncogene Tu which shows a relation to a cellular counterpart of the transforming src oncogene of avian sarcoma virus, the c-src [30]. The normal function of c-src remained unknown. Since c-src, however, was also found in all individuals ofall metazoans tested, and was not found in protozoans and plants, it might have some basic functions of life closely related to the multicellular organization of animals including humans [36]. In Xiphophorus it was shown further that the cellular oncogene is normally under control of systems of multiple regulatory genes corresponding to regulator genes of bacteria and phages. Some of the regulatory genes are located on other chromosomes than those bearing an oncogene. One of the most prominent regulatory genes appears to be responsible for terminal differentiation of the neoplastically transformed cells exerted via modification of nucleosides in the anticodon of certain tRNAs [40]. Interpopulational or interracial hybridization in preceding generations in Xiphophorus is the main event contributing to the disintegration of the regulatory gene system for the oncogene. Germ line mutations that may also disturb the regulatory gene systems are probably less important than hybridization because they are always rare, or may become repaired. Somatic mutations and tumor promotion, which are the majority of carcinogenic triggers, may complete this disintegration. The majority of the neoplasms of Xiphophorus belongs to the types that are triggered by carcinogens or promoters on a competent genetic background like their counterparts in humans, which represent about 90% of all human neoplasms (see [8]). The phenomenon of introducing susceptibility to neoplasia by means of hybridization is not limited to Xiphophorus. Many examples have been cited from the animal kingdom [8]. It appears that in animals from wild populations neoplasia is difficult to induce and "spontaneously" developing neoplasms are rare, while in animals of hybrid origin ( domesticated and laboratory animals; naturally occurring and experimentally produced hybrids) neoplasia is easily inducible and the incidence of "spontaneously" developing neoplasms is high. While we do not have hybridization in human beings comparable to hybridization of domesticated or laboratory animals such as fish and mice, it is suggestive to speculate how much effect hybridization may have had on the high tumor incidence observable in some of our highly developed nations. Such speculations are probably of little value in the fight against cancer, but in our search for the cause of human neoplasia they could help to realize the factors that make an individual susceptible to neoplasia and, therefore, sensitive to the carcinogens of our environment.
I am grateful to Dr. Heinz Bailer (Giessen) and Dr. Helga Kersten (Erlangen) for their design and realization of the crucial experiments recently performed in their laboratories as well as for the so far unpublished data included in this review. Thanks are also due to Dr. Nishimura (Tokyo) for providing the laboratory of Dr. Kersten with the insertase of E. coli. The critical discussion and assistance of Dr. M. Schartl, Dr. E. Scholl, Dr. A. Barnekow, and Dr. A. Anders in preparing the manuscript are gratefully acknowledged. The research was supported by Deutsche Forschungsgemeinschaft through Sonderforschungsbereich 103 "Zellenergetik und Zelldifferenzierung", Marburg, Stiftung Volkswagenwerk, Bundesministerium für Forschung und Technologie, and by Land Hessen through Justus-Liebig-Universität Giessen.
1. Kallman K (1975) In: King RC (ed) Handbook of Genetics, vol 4, p 81, Plen ilm Press, New York 2. Anders A, Anders F ( 1978) Biochim Biophys Acta 516:61 3. Rosen O (1979) Bull Amer Nat Hist 162:267 4. Schwab M (1980) Verh Otsch Zool Ges 73:285 5. Scholl A, Anders F (1973) Archiv für Genetik 46: 121 6. Scholl E ( 1980) Thesis, Giessen 7. Kollinger G, Siegmund E (1981) Verh Otsch Zool Ges 74: 206 8. Anders F, Schartl M, Scholl E ( 1981) In : Oawe C et al. (eds) Phyletic Approaches to Cancer. Japan Sci Soc Press, Tokyo, p 189 9. Schwab M, Haas J, Abdo S, Ahuja MR, Kol linger G, Anders A, Anders F (1978) Ex perientia 34:780 10. Schwab M, Abdo S, Ahilja MR, Kollinger G, Anders A, Anders F, Frese K (1978) Z Krebsforsch 91: 301 11. Prescott DM, Flexer AS (1982) Cancer: The Misguided Cell, Sinauer Associate Inc. Publishers, Sunderland, Mass 12. Heston WE (1974) Heredity 65:262 13. Anders A, Anders F, Kline K (1973) In: Schröder JH (ed) Genetics and Mutagenesis of Fish, I 33, II 53, Springer, Berlin Heidelberg New York 14. Ahilja MR, Schwab M, Anders F (1980) J of Heredity 71:403 15. Siciliano MJ, Wright OA (1976) Prog Exp Tumor Res 20: 398 16. Anders F, Schwab M, Scholl E (1981) In: Stich HF, San R (eds) Short Term Tests for Chemical Carcinogens, Springer, Berlin Heidelberg New York, p 399 17. Vielkind J, Vielkind U, Anders F (1971) Z Krebsforsch 75: 243 18. Vielkind U, Schlage W, Anders F (1977) Z Krebsforsch 90:285 19. Anders F, Oiehl H, Schwab M, Anders A (1979) In: Klaus SN (ed) Pigmentation, its Genesis and Biological Control, vo14, p 142 20. Anders F, Oiehl H, Scholl E ( 1980) In: Spearman RIC, Riley PA (eds) The Skin of Vertrebrates, Linnean Society Symposium Series Number 9. Academic Press, London p211 21. Schartl M, Schartl A, Anders A (1981) In: Seiji M ( ed) Pigment Cell, University of Tokyo Press, p 507 22. Ahuja MR, Lepper K, Anders F (1979) Ex perien tia 35: 28 23. Anders F, Klinke K (1966) Verh Dtsch Zool Ges 30: 391 24. Schwab M, Vielkind J, Anders F (1976) Mol Gen Genet 144: 151 25. Vielkind J, Haas-Andela H, Vielkind U, Anders F (1982) Mol Gen Genet 185:379 26. Wigler M, Pellicer A, Silverstein S, Axel R (1978) Ce1114:725 27. Willecke K (1980) In: Celis JE, Graessmann A, Loyter A (eds) Transfer of Cell Constituents into Eukaryotic Cells. Plenum Press, New York, London, p 311 28. Stehelin D, Varmus HE, Bishop JM (1976) Nature 260: 170 29. Spector DH, Varmus HE, Bishop JM (1978) Proc Natl Acad Sci Wash 75:4102 30. Bauer H, 33. Kolloquium Mosbach 1982. (in press) 31. Barnekow A, Schartl M, Anders F, Bauer H (1982) Cancer Res 42:2429 32. Czernofsky AP, Schartl M (unpublished) 33. Collett MS, Purchio AF, Erikson RL (1980) Nature285: 167 34. Schartl M, Barnekow A, Bauer H, Anders F (1982) Cancer Res 42:4222 35. Chatterjee K, Kollinger G, Schmidt R-C, Anders A, Anders F (1981) Cancer Genetics and Cytogenetics 3: 195 36. Schartl M, Bamekow A (unpublished) 37. Shilo BZ, Weinberg RA (1981) Proc Natl Acad Sci Wash 78:6789 38. Vielkind U (1976) J Exp Zoo1196: 197 39. Schartl M (1979) Thesis, Giessen 40. Kersten H, 33. Colloquium Mosbach 1982. (in press ) 41. Okada N, Shindo-Okada N, Sato Sh, Itoh YH, Oda K-I, Nishimura S (1978) Proc Natl Acad Sci Wash 75:4247 42. Kersten H (1982) In: Usdin E, Borchardt R, Greveling B ( eds) Elsevier North-Holland Inc (in press) 43. Kersten H (in press) J of Cancer Res and Clinical Oncology 44. Shindo-lkada N, Terada M, Nishimura S (1981) Eur J Biochem 115:423 45. Kasai H, Kuchino Y, Nhei K, Nishimura S (1975) Nucl Acids Res 2: 1931 46. Dess G (1982) Thesis, Giessen |
