|
Department of Medicine, Box 420, The University of Chicago, 950 East 59th Street, Chicago. Illinois 60637, USA
The renewed interest in the study of chromosome abnormalities in hematologic malignancies, particularly in the leukemias, is the result of technical improvements which permit the precise identification of each human chromosome, and of parts of chromosomes as well. The information obtained raises a number of questions regarding the validity of older notions, such as the variability of the chromosome pattern (karyotype) in acute leukemia, or the rarity of associations of specific chromosome abnormalities with particular types of leukemia. One of the surprising observations of the last few years has been the frequent occurrence of consistent translocations in a variety of hematologic malignancies. The challenging questions at present are how and why nonrandom changes, particularly consistent translocations, occur.
An analysis of chromosome patterns in malignancy must be based on a study of the karyotype of the tumor tissue itself. In the case of leukemia, the specimen is usually a bone marrow aspirate that is processed immediately or is cultured for a short time [29]. Cells in metaphase from a 24-hour culture of peripheral blood will have a karyotype similar to that of cells obtained from the bone marrow. The chromosome analysis may be performed by means of one of several pretreatments prior to staining with Giemsa [34 ], or the slides can be stained with quinacrine mustard for fluorescence, as previously described [3,29]. The chromosomes are identified according to the Paris Nomenclature [22], and the karyotypes are expressed as recommended under this system.
I. Chronic Phase Table I. The most frequent chromosome
changes determined with banding in 136 Ph1-positive 
The identity of the cells that contain the PhI chromosome has recently become a topic of considerable interest. This problem has at least two facets; one concerns the nature of the blast cells in the acute phase of CML and the other, the proper classification of patients with PhI + acute leukemia. In regard to the first aspect, Boggs [1} noted that the blast cells in some patients in the acute phase of CML appeared to be lymphoid rather than myeloid, and that some patients in the acute phase achieved a remission with vincristine and prednisone, which were usually effective primarily in lymphoid leukemias. Severallaboratories are currently examining the surface markers of cells from patients in the acute phase of CML; unfortunately. the cytogenetic analyses are frequently not done with banding techniques, and often the karyotype is obtained only from the initial sample. Since Whang-Peng et al. [39] have identified the PhI chromosome in two of four PhI + ALL patients as a 21q -, banding is essential. In regard to the second question, of 13 patients [26] with PhI+ ALL who had a 22q -chromosome identified with banding, six had a translocation of 22q to 9q34; two others had variant translocations, one to 14q and one to 21q. The presence of a translocation was not determined for the other five. Ten of the 13 patients were studied a second time; two of these had no remission and continued to have an abnormal karyotype. The remaining eight patients achieved a remission and had a normal karyotype in cells from the bone marrow or from unstimulated peripheral blood. It remains to be determined whether it is logical, or correct, to classify all PhI + leukemias as CML, or whether we are dealing with two different diseases.
I. Nonrandom Patterns 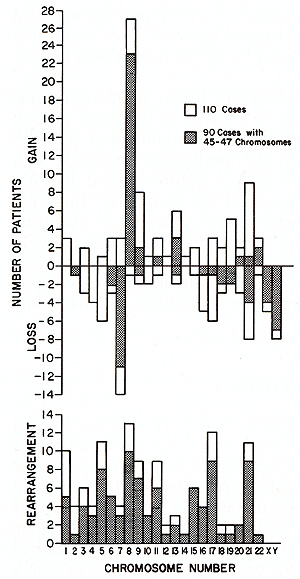
The mechanism for the production of specific, consistent reciprocal translocations is unknown. Possibly, specific translocations are the result of cell selection. In such a model, chromosome breaks and rearrangements occur continuously at a low frequency. Many of these rearrangements do not lead to changes in cell metabolism, and the cells therefore do not proliferate preferentially; other rearrangements may be lethal to the cells. Still others provide the cell with a proliferative advantage, and cells with these changes not only persist, but eventually become the predominant cell type. In such a model, the chromosome change is the fundamental, initial event that leads to the neoplastic nature of the cell. Other possible explanations depend on either [I] chromosome proximity, since translocations may occur more frequently when two chromosomes are close together, or [2] regions of homologous DNA that might pair preferentially and then be involved in rearrangements. The fact that many of the affected chromosomes, e.g., Nos. 1,9, 13, 14, 15,21, and 22, are involved in nucleolar organization supports these proposals. On the other hand, proximity of homologous DNA sequences should lead to an increased frequenced of these rearrangements in patients with constitutional abnormalities, but this has not been observed. It is possible that either or both of these mechanisms are subject to selection; a translocation might occur because the chromosomes are close together, but only certain specific rearrangements may have a proliferative advantage which results in leukemia and thus leads to their detection. Another genetic mechanism that may account for consistent chromosome changes is related to transposable elements, called controlling elements in maize [6, 15] and insertion sequences in bacteria [4]. Transposable elements have been detected in every organism in which the genetic structure is known with reasonable precision. In maize, for example, there are at least three distinct controlling elements, each with its own characteristics and with different chromosome locations that influence the production of anthocyanin pigment in each kernel of an ear of corn [6, 15]. Similar genetic systems that modify the action of host genes may be present in mammalian cells. If so, these transposable elements may playa role in malignant transformation. The following features of transposable elements are relevant to the "how" and "why" of consistent translocations: 1. Change in location within the DNA, 2. the transferring of adjacent DNA in this change, and 3. the alteration of the normal mechanism for genetic regulation, depending on the site and orientation of the inserted sequences. These properties, plus a selective system for removal of changes that do not have a proliferative advantage in hematologic cells, are just those required to explain consistent translocations occurring as somatic mutations.
There is good cytological [8] and biochemical [5] evidence that, in an individual patient with chronic myelogenous leukemia or Burkitt lymphoma, the tumor cells have a clonal origin. In CML, initially only a single cell has the 9: 22 translocation, and when the patient comes to the physician, frequently all cells in division contain the PhI chromosome. It is necessary to examine the kinds of genetic mechanisms that can provide the cell containing the 9: 22 translocation with this proliferative advantage. Two points that should be emphasized are the genetic heterogeneity of the human population and the variety of cells involved in malignancy. There is convincing evidence from animal experiments that the genetic constitution of an inbred strain of rats or mice plays a critical role in the frequency and type of malignancies that develop [23,33]. We are much more aware now than formerly of certain genes in man that predispose to cancer, such as the genes for Bloom syndrome, Fanconi anemia, and ataxia-telangiectasia [9]. We are completely ignorant of the number of gene loci in man which. in some way. control resistance or susceptibility to a particular malignancy. The second factor affecting the karyotypic pattern relates to the different cells that are at risk of becoming malignant, and the varying states of maturation of these cells. There is good evidence that the same chromosomes may be affected in a variety of tumors; No.8 is a good example [ 18]. On the other hand, some chromosomes seem to be involved in neoplasia involving a particular tissue: the involvement of No. 14 in lymphoid malignancies is an example. When one considers the number of nonrandom changes that are seen in a single malignancy such as ANLL, it is clear that not just one gene, but rather a class of genes is involved. Our knowledge of the human gene map [ 17] has developed concurrently with our understanding of chromosome changes in leukemia. It is now possible to try to correlate the affected chromosomes with the genes that they carry. Clearly, these efforts must be very tentative. since relatively few genes have been mapped, and since some of the chromosomes that are most frequently abnormal have few genetic markers. Preliminary data suggest that chromosomes which carry genes related to nucleic acid biosynthesis may frequently be abnormal in hematologic malignancies. Moreover, specific chromosome regions associated with these genes may also be involved. Thus, the most frequent abnormalities of No. 17 result either in an isochromosome for the long arm or in a translocation with No. 15 in which the break in No. 17 is in band 17q22. This region of No. 17 contains genes for thymidine kinase, galactokinase, and a site that is particularly vulnerable to AD-12-induced breakage [16]. Furthermore, induction of host cell thymidine kinase and a high frequency of breaks in 17q22 are early functions of this virus, as is the synthesis of a tumor antigen which may playa role in the control of DNA synthesis. Thus it is possible that nonrandom chromosome aberrations, when they occur. change the level of some enzymes related to nucleic acid metabolism, . either through a change in location or through duplication of gene loci. Nonrandom chromosome changes, particularly consistent, specific translocations. now seem clearly to be an important component in the proliferative advantage gained by the mutant cell in neoplasia. The challenge is to decipher the meaning of these changes. G. Summary The consistent occurrence of nonrandom chromosome changes in human malignancies suggests that they are not trivial epiphenomena. Whereas we do not understand their significance at present, one possible role which they may fulfill is to provide the chromosomally aberrant cells with a proliferative advantage as the result of alteration in the number or location of genes related to nucleic acid biosynthesis. The proliferative advantage provided by various chromosome aberrations is likely to differ in patients with different genetic constitutions.
Supported by the National Foundation -March of Dimes. the National
Institutes of Health (CA 16910). the Leukemia Research Foundation.
I. Boggs. D. R.: Hematopoietic stem cell theory in relation to possible lymphoblastic conversion or chronic myeloid leukemia. Blood 44,449-453 (1974) 2. Caspersson, T., Gahrton, G., Lindsten, I., Zech, L.: Identification of the Philadelphia chromosome as a number 22 by quinacrine mustard fluorescence analysis. Exp. Cell Res. 63,238-244 (1970a) 3. Caspersson, T., Zech, L., Iohansson, C., Modest, E.I.: Identification of human chromo somes by DNA-binding fluorescent agents. Chromosoma 30,215-227 (1970b) 4. Cohen, S. N.: Transposable genetic elements and plasmid evolution. Nature 263, 731-738 (1976) 5. Fialkow, P.I.: The origin and development of human tumors studied with cell markers. N. Engl. I. Med. 291,26-35 (1974) 6. Fincham, I.R.S., Sastry, G.R.K.: Controlling elements in maize. Ann. Rev. Genet. 8, 15-50 (1974) 7. First International Workshop on Chromosomes in Leukemia: Chromosomes in acute non lymphocytic leukemia. Brit. I. Haematol., 39,311-316 (1978) 8. Gahrton, G., Lindsten, I., Zech, L.: Clonal origin of the Philadelphia chromosome from either the paternal or the maternal chromosome number 22, Blood 43,837-840 (1974) 9. German, I.: Genes which increase chromosomal instability in somatic cells and predispose to cancer. In: Progress in Medical Genetics VIII, Steinberg, A.G., Beam. A.G. (eds.). pp, 61-101, New York: Grune & Stratton 1972 10. Golomb, H. M., Rowley, I. D" Vardiman, I., Baron. I" Locker, G,. Krasnow. S.: Partial deletion of long arm of chromosome 17. Arch. Intern. Med, 136, 825-828 ( 1976) 11. Golomb, H. M., Vardiman, J., Rowley, I. D.: Acute non-lymphocytic leukemia in adults: Correlations with Q-banded chromosomes. Blood 48,9-21 (1976) 12. Golomb, H. M., Vardiman, J.W., Rowley, J. D., Testa, J. R., Mintz, U.: Correlation of clin ical findings with quinarine-banded chromosomes in 90 adults with acute nonlympho cy tic leukemia. New England I. Medicine 299,613-619 (1978) 13. Ishihara, T., Kohno, S.-I., Kumatori, T,: Ph1-translocation involving chromosome 21 and 22. Br. J. Cancer 29,340-342 (1974) 14. Mayall, B. H., Carrano, A.V., Moore. D. H. II, Rowley. I, D.: Quantification by DNA-based cytophotometry of the 9q + /22q -chromosomal translocation associated with chronic myelogenous leukemia, Cancer Res. 37,3590-3593 (1977) 15. McClintock, B.: The control of gene action in maize, In: Genetic Control of Differentiation. Brookhaven Symp. Biol. 18, 162-184 (1965) 16. McDougall, I. K., Kucherlapati, R. S., Ruddle. F. H.: Localization and induction of the human thymidine kinase gene by adenovirus 12, Nature (New BioI.) 245,172-175 (1973) 17. McKusick, V, A., Ruddle, F. H. : The status of the gene map of the human chromosomes. Science 196, 390-405 ( 1977) 18. Mitelman, F., Levan, G.: Clustering of aberrations to specific chromosomes in human neo plasms. Hereditas 82, 167-174 (1976) 19. Nilsson, P.G., Brandt, L., Mitelman, F.: Prognostic implications of chromosome analysis in acute non-lymphocytic leukemia. Leukemia Research 1,31-34 ( 1977) 20. Nowell, P,C., Hungerford. D.A.: A minute chromosome in human chronic granulocytic leukemia. Science 132, 1197 ( 1960) 21. O'Riordan, M.L., Robinson. I,A" Buckton. K.E.. Evans, H.J.: Distinguishing between the chromosomes involved in Down's syndrom (trisomy 21) and chronic myeloid leukemia (Phl) by fluorescence, Nature 243,167-168 (1971) 22. Paris Conference 1971: Standardization in human cytogenetics. In: Birth Defects. Original Article Series, VIII: 7. New York: The National Foundation 1972 23. Rowe, W, P.: Genetic factors in the natural history of murine leukemia virus infection. Cancer Res. 33,3061-3068 (1973) 24, Rowley, I. D.: A new consistent chromosomal abnormality in chronic myelogenous leukemia identified by quinacrine fluorescence and Giemsa staining. Nature (Lond.) 243, 290-293 (1973a) 25. Rowley, I.D.: Identification of a translocation with quinacrine fluorescence in a patient with acute leukemia. Ann. Genet. 16,109-112 (1973b) 26. Rowley, I. D.: The cytogenetics of acute leukemia. Clin. Haematol. 7,385-406 (1978a) 27, Rowley, I. D. : Chromosomes in leukemia and lymphoma. Seminars in Hematology, 15, 301-319 (1978b) 28. Rowley, J. D.: Chromosome abnormalities in the acute phase of CML. Virchows Arch. B Cell Path. 29,57-63 (1978c) 29. Rowley, J.D., Potter, D.: Chromosomal banding patterns in acute non-lymphocytic leukemia. Blood 47,705-721 (1976) 30. Rowley, J. D., Golomb, H. M., Vardiman, J., Fukuhara, S., Dougherty, C., Potter, D.: Further evidence for a non-random chromosomal abnormality in acute promyelocytic leu kemia. Int. J. Cancer 20,869-872 (1977) 31. Sakurai. M.. Sandberg. A.A.: XI. Correlations of karyotypes with clinical features of acute myeloblastic leukemia. Cancer 37,285-299 ( 1976) 32. Sonta, S.. Sandberg. A. A.: XXIV. Unusual and complex Ph1 translocations and their clini cal significance. Blood 50,691-697 ( 1977) 33. Steeves, R., Lilly. F.: Interactions between host and viral genomes in mouse leukemia. Ann. Rev. Genet. 11,277-296 (1977) 34. Sumner. A.T.. Evans. H.J.. Buckland. R.A.: New techniques for distinguishing human chromosomes. Nature (New BioI.) 232,31-32 (1971) 35. Testa. J. R.. Rowley. J. D.. Mintz, U.. Golomb. H. M.: Evolution of karyotypes in acute non lymphocytic leukemia (ANLL). Am. Sociol. Hum. Genet.. 95A (1978) 36. Testa. J. R.. Rowley. J. D.: Cytogenetic patterns in acute nonlymphoblastic leukemia. Virchows Arch. B Cell Pa th .29. 65-72 ( 1978 ) 37.Trujillo. J.M.. Cork. A., HartJ.S.. George.S.L.. Friereich. E.J.: Clinical implications of aneuploid cytogenetic profiles in adult acute leukemia. Cancer 33,824-834 ( 1974) 38. Whang-Peng. J.. Canellos. G. P.. Carbone. P. P.. Tjio. J. H.: Clinical implications or cyto genetic variants in chronic myelocytic leukemia (CML). Blood 32,755-766 ( 1968) 39. Whang-Peng. J., Knutsen. T., Ziegler. J.. Leventhal. B.: Cytogenetic studies in acute lymphocytic leukemia: Special emphasis in long-term survival. Med. Pediatr. Oncol. 2, 333-351 ( 1976) |