|
1 Department of Genetics, Weizmann Institute of Science, Rehovot 76100, Israel A. Cloning and Clonal Differentiation of Normal Hematopoietic Cells in Culture The cloning and clonal differentiation of normal hematopoietic cells in culture made it possible to study the controls that regulate growth (multiplication) and differentiation of different hematopoietic cell types; see [63-68]. We first showed [17,59], as was then confirmed by others [4], that normal mouse myeloid precursor cells cultured with a feeder layer of other cell types can form clones of granulocytes and macrophages in culture. We also found that the formation of these clones is due to secretion by cells of the feeder layer of specific inducers that induce the formation of clones and the differentiation of cells in these clones to macrophages or granulocytes in mice [26, 59, 60] and in humans [57]. After we first detected their presence in culture supernatants [26, 60], these protein inducers have been referred to by a number of names and I shall use the name macrophage and granulocyte inducers (MGI) (Table I). These proteins can be produced and secreted by various normal and malignant cells in culture and in vivo [63]. Their production can be induced by a variety of compounds [10, 12, 41, 80] and some cells produce these proteins constitutively [ 1, 26, 31, 34, 71]. MGI are a family of proteins that exist in a number of molecular forms that have different biologic activities. This cell culture approach has led to the cloning and isolation of growth factors for all the different types of hematopoietic cells, including different types of lymphocytes.
The family of MGI proteins include some proteins that induce cell growth (multiplication) and others that induce differentiation. Those that induce growth, which are also required for normal cell viability, we now call MGI-l. These include proteins that induce the formation of macrophage clones (MGI-IM) [26, 48, 71], granulocyte clones (MGI-IG) [26,48,54], or both types of clones (MGI-IGM) [6, 31, 34]. MGI-l has previously been referred to as mashran gm [27], colony-stimulating factor (CSF) [51], colony-stimulating activity (CSA) [1], and MGI [31] (Table 1 ). The existence of an antibody that does not react with all forms of MGI-IM or MGI-IG has shown that there can be different antigenic sites on molecules that belong to the same form of MGI-l [47, 48]. The other main type of MGI, which we now call MGI-2 [33, 48, 66], induces the differentiation of myeloid precursor cells, either leukemic [14] or normal [33, 66], without inducing colony formation. This differentiation-inducing protein [13, 14] has also been referred to as MGI [14], D factor [49, 82], and GM-DF [5]. It has been suggested that there are different forms of MGI-2 for differentiation to macrophages or granulocytes [33]. The regulation of MGI-l and MGI-2 appears to be Table I. In vitro cloning and clonal
differentiation of normal hematopoietic cells 
Normal myeloid precursor cells isolated from bone marrow [37] require
an external source of MGI-l for cell viability and growth. There
are, however, myeloid leukemic cells that no longer require MGI-l
for viability and growth, so that these leukemic cells can then
multiply in the absence of MGI-l [64, 66]. This gives the leukemic
cells a growth advantage over the normal cells when there is a limiting
amount of MGI-l. Starting with a decreased requirement for MGI-l,
this eventually leads to a complete loss of this requirement. Other
myeloid leukemic cells constitutively produce their own MGI-l [54,
56] and these leukemic cells also have a growth advantage compared
with normal cells that require an external source of MGI-l (Fig.
I). A change in the requirement of MGI-l for growth, either a partial
or complete loss of this requirement, or the constitutive production
of MGI-l, thus both give a growth advantage to leukemic cells. The
existence of myeloid leukemic cells that either no longer require
MGI-l for viability and growth or constitutively produce their own
MGI-l, raises the question 
whether these leukemic cells can still be induced to differentiate to mature cells by the normal differentiation-inducing protein MGI-2. This question has been answered by showing that there are clones of myeloid leukemic cells that no longer require MGIl for growth, but can still be induced to differentiate normally to mature macrophages and granulocytes by MOI-2 via the normal sequence of gene expression; see [64-68]. These mature cells are then no longer malignant in vivo [ II, 43, 47]. Injection of these myeloid leukemic cells into embryos has shown that after such injection the leukemic cells can participate in hematopoietic differentiation in apparently healthy adult animals [18,78]. Injection of MGI-2 into animals, or in vivo induction of MGI-2 by a compound that induces the production of this differentiation-inducing protein, results in an inhibition of leukemia development in animals with such leukemic cells [43, 47]. There are also myeloid leukemic cells that constitutively produce their own MGI-l and that can be induced to differentiate by MGI-2. Our results indicate that induction of normal differentiation in myeloid leukemic cells by MGI-2 can be an approach to therapy based on the induction of normal differentiation in malignant cells [ 14, 40, 43, 46, 47, 57]. There are various forms of MGI-2 which differ in their ability to induce differentiation in different clones of myeloid leukemic cells [40,43,46,47]. Leukemic clones that can be induced to differentiate to mature cells by MOI-2 have been found in different strains of mice [ 5, 14, 15, 25, 28, 38]. They are referred to as MGI+D+ (MGI+ to indicate that they can be induced to differentiate by MGI-2; D+ for differentiation to mature cells). MGI+D+ leukemic cells have specific chromosome changes compared with normal cells [2, 19]. These chromosome changes thus seem to involve changes in genes other than those involved in the induction of normal differentiation. There are other clones of myeloid leukemic cells that can also grow without adding Mal-l, but that are either partly (MGI+D-) or almost completely (MGI-D-) blocked in their ability to be induced to differentiate by MOI-2 [15,21, 23, 28, 48, 69, 70]. These differentiation-defective clones have specific chromosome changes compared with MOI+D+ cells [2, 19]. There are a variety of compounds, other than MGI-2, that can induce differentiation in MGI+D+ clones. Not all these compounds are active on the same MGI+D+ clone, and they do not all induce the same differentiation-associated properties. The inducers include certain steroids, lectins, polycyclic hydrocarbons, tumor promoters, lipopolysaccharides, X-irradiation, and compounds used in cancer chemotherapy [42, 64]. The existence of clonal differences in the ability of X-irradiation and cancer chemotherapeutic chemicals to induce differentiation may help to explain differences in response to therapy in different individuals [64]. As a result of these experiments, we have suggested that it may be possible to introduce a form of therapy based on induction of differentiation [14,40,42,43,57, 63-65]. This would include prescreening in culture to select for the most effective compounds, and using these compounds for a low dose chemotherapy protocol aimed at inducing cell differentiation [42]. Since different myeloid leukemic clones respond differently to MGI-2 and other compounds, such differences will also occur in leukemic cells from different patients. Based on these suggestions [63, 64], some encouraging clinical results have been obtained with the use of low dose cytosine arabinoside [3, 24, 52].
Some of the compounds that induce differentiation in susceptible clones of MGI-D+ leukemic cells, including lipopolysaccharide, phorbol ester tumor promoters such as 12-0-tetradecanoylphorbol-13-acetate (TP A), and nitrosoguanide, can induce the production of MGI-2 in these clones. These compounds thus induce differentiation by inducing in the leukemic cells the endogenous production of the normal differentiation-inducing protein MGI-2 [10,41,80]. Other compounds such as the steroid dexamethasone, can induce differentiation in MGI+D+ clones without inducing MGI-2 [10]. This steroid induces differentiation by other pathways of gene expression than MGI-2 [7, 38]. The same applies to dimethylsulfoxide (DMSO). Induction of differentiation in some myeloid leukemic clones requires combined treatment with different compounds [30, 39, 41, 74, 75]. In these cases, one compound induces changes not induced by the other, so that the combined treatment results in new gene expression. This complementation of gene expression can occur both at the level of mRNA production and mRNA translation [22]. With the appropriate combination of compounds, we have been able to induce all our MGI-D- leukemic clones for some differentiation-associated properties [74, 75]. It will be interesting to determine whether the same applies to differentiation of erythroleukemic cells [16, 50]. It is possible that all myeloid leukemic cells no longer susceptible to the normal differentiation-inducing protein MGI-2 by itself, can be induced to differentiate by choosing the appropriate combination of compounds to give the required complementation. This can include the use of hormones such as steroids [35, 36], or insulin [73, 74], and different nonphysiologic compounds [64], with or without MGI-2.
We have developed a simple procedure for isolating normal myeloid precursor cells from the bone marrow [37]. Incubation of isolated normal myeloid precursors with MGI-l, either MGI-IM or MGI-IG [48], induces the viability and growth of these normal precursors, and results in cell differentiation to macrophages or granulocytes, even without adding the differentiation-inducing protein MGI-2. The incubation of normal myeloid precursors with MGI-l also results in the induction of MGI-2 [33,44, 45,66]. This induction of MGI-2 can be detected as early as 6 h after the addition of MGI-l [44]. This induction of MGI-2 by MGI-l can thus account for the induction of differentiation after adding MGI-I to the normal cells. The induction of differentiation-inducing protein MGI-2 by growthinducing protein MGI-I thus appears to be an effective control mechanism for coupling growth and differentiation in the normal cells. It has been shown that the receptor for epidermal growth factor has tyrosine-specific protein kinase activity [76]. This has also been found for receptors for other growth factors such as insulin [29] and presumably also applies to the receptor for the myeloid cell growth-inducing protein MGII. The myeloid differentiation-inducing protein MGI-2, but not MGI-l, can bind to cellular DNA [79]. This shows that growth and differentiation in normal myeloid cells are coupled by induction of a differentiation-inducing, DNA-binding protein by a growth-inducing protein. This mechanism for coupling growth and differentiation may also apply to other types of cells. Differences in the time of the switch-on of the differentiation inducer would produce differences in the amount of multiplication before differentiation. The platelet-derived growth factor is structurally related to the simian sarcoma virus oncogene sis [9,77]. It will be interesting to determine whether MGI-l and MGI-2 are structurally related to any of the known oncogenes. The multiplication of normal cells is regulated at two control points. The first control is that which requires MGI-l to produce more cells that can then differentiate by the MGI-2 induced by MGI-l. The second control is the stopping of cell multiplication that occurs as part of the program of terminal differentiation to mature cells induced by MGI-2. There is thus a coupling of growth and differentiation in normal cells at both these points.
As pointed out already, there are MGI+D+ clones of myeloid leukemic
cells that no longer require MGI-l for growth, but can still be
induced to differentiate normally by MGI-2. These leukemic cells
have thus uncoupled the normal requirement for growth from the normal
requirement for differentiation. Experiments on the properties of
these cells after induction of differentiation by MGI-2 have shown
that the normal requirement for MGI-l for cell viability and growth
is restored in the differentiating leukemic cells [13, 44, 45].
MGI-1 added to normal myeloid precursors induces the production
of MGI-2, so that the cells can then differentiate by the endogenously
produced MGI-2. However, in these leukemic cells, MGI-l did not
induce the production of MGI-2 even though, like normal cells, they
again required Mal-l for viability and growth. There was therefore
no induction of differentiation after adding MGI-l [44, 45]. There
is another type of leukemic cell that constitutively produces its
own MGI-l and can also show this lack of induction of MGI-2 by MGI-l,
so that the cells do not differentiate [72]. The absence of induction
of MGI-2 by MGI-l therefore uncouples growth and differentiation
in these leukemic cells. The lack of requirement of Mal-l for growth
and the absence of induction of the differentiationinducing protein
MaI-2 by the growth-inducing protein MGI-l, are thus mechanisms
that uncouple growth and differentiation in MGI+D+ leukemic cells
[44, 45, 66, 72]. In leukemic cells with constitutive production
of MGI-l, changes in specific components of the culture medium can
result in an autoinduction of differentiation owing to the restoration
of the induction of Mal2 by MGI-l, which then restores the normal
coupling of growth and differentiation (Fig.2). These changes in
the culture medium include the use of mouse or rat serum instead
of horse or calf serum, serurn-free medium, and removal of transferrin
from serum-free medium [72]. Autoinduction of differentiation in
this type of leukemic cell may also occur under certain conditions
in VIVO. This coupling of growth and differentiation in normal cells
is regulated at two control points. The uncoupling of growth and
differentiation in MGl+D+ leukemic cells is at the first control
point, but the coupling at the second control in normal cells, between
the induction of differentiation by MGI-2 and the stopping of multiplication
in the mature cells, is maintained. There are differentiation-defective
MGI+D- leukemic cells, that, like the MGI+D+ leukemic cells, no
longer require addition of MGI-l for growth. However, in these cells
MGI-2 induces only a partial differentiation, mature cells are not
produced, and the cells do not stop multiplying. In addition to
uncoupling growth and differentiation at the first control point,
MGI+D-leukemic cells thus show a second uncoupling between the initiation
of differentiation by MGI-2 and the stopping of cell multiplication
that occurs as part of the normal program of terminal differentiation.
It has been suggested that leukemia originates by uncoupling the
first control and that uncoupling of the second control then results
in a further evolution of leukemia [64, 66]. 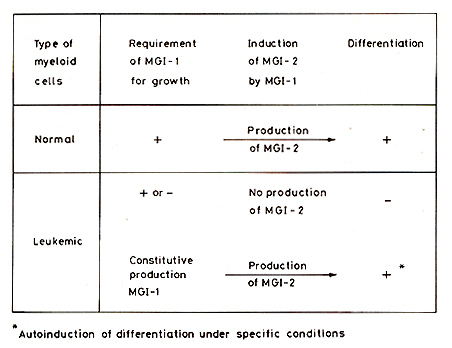
Since there are leukemic cells which, unlike normal myeloblasts, no longer require MGI-l for cell viability and growth, the molecular changes required for viability and growth that have to be induced in the normal cells are constitutive in these leukemic cells. This also applies to leukemic cells that constitutively produce their own MGI-l. This suggests that the origin of myeloid leukemia can be due to a change from an induced to a constitutive expression of genes that control cell viability and growth [64,66]. Studies on changes in the synthesis of specific proteins in normal myeloblasts, MGI+D+, MGI+D-, and MGI-D-leukemic clones at different times after adding MGI-land MGI-2, using two-dimensional gel electrophoresis [32], have directly shown that there have been changes from inducible to constitutive gene expression in the leukemic cells. The results also indicate a relationship between constitutive gene expression and uncoupling of the induction of differentiation by MGI-2 and the stopping of multiplication in the mature cells. The results indicate that changes from an induced to a constitutive expression of certain genes are associated with the uncoupling of growth and differentiation, both at the control which requires MGI-l to produce more cells and at the control of the stopping of cell multiplication that occurs in the formation of mature cells. The protein changes during the growth and differentiation of normal myeloblasts seem to be induced by MGI-l and MGI-2 as a series of parallel multiple pathways of gene expression [32]. It can be assumed that the normal developmental program that couples growth and differentiation in normal cells requires synchronous initiation and progression of these multiple parallel pathways. The presence of constitutive gene expression for some pathways can be expected to produce asynchrony in the coordination required for the normal development program. Depending on the pathways involved, this asynchrony could then result in an uncoupling of the controls for growth and differentiation and produce different blocks in the ability to be induced for the differentiation process and to terminate it. We have been able to treat MGI-O- leukemic cells so as to induce the reversion of specific proteins from the constitutive to the nonconstitutive state. This reversion was then associated with a gain of inducibility by MGI-2 for various differentiationassociated properties. Reversion from the constitutive to the nonconstitutive state in these cells thus restored the synchrony required for induction of differentiation [75]. The suggestion derived from these results [32, 64, 66] is, therefore, that myeloid leukemia originates by a change that produces certain constitutive pathways of gene expression, so that cells no longer require Mal-l for growth or constitutively produce Mal-l without inducing MGI-2. These leukemic cells can, however, still be induced to differentiate normally by MGI-2 added exogenously or induced in the cells in other ways. The differentiation program induced by MGI-2 can thus proceed normally when it is uncoupled from the growth program induced by Mal-l. This can be followed by constitutive expression of other pathways, resulting in the uncoupling of other controls and an asynchrony that interferes with the normal program of terminal differentiation. These second changes then result in the further evolution of leukemia [66].
These conclusions on the origln and evolution of myeloid leukemia may be applicable to malignant tumors derived from other types of cells whose via bility , growth, and differentiation are induced by other physiologic inducers. Identification of the physiologic inducers of growth and differentiation for different cell types would be a crucial requirement in extending these conclusions to those other tumors. However , even in the absence of such identifications, it appears likely that teratocarcinoma cells [8, 53] may be comparable to MGI+D+ myeloid leukemic cells. The presence of fetal proteins in certain tumors may also be due to constitutive gene expression in the tumor of a protein that is induced by the physiologic inducer during the developmental program in the normal fetus [66]. There are probably a variety of tumors in which: (a) the original malignancy has a normal differentiation program and the cells are malignant because of uncoupling of the requirement for growth from the requirement for differentiation by changing the gene expression required for growth from inducible to constitutive; and (b) where the further evolution of the tumor results from changes from inducible to constitutive of other pathways of gene expression that produce asynchrony in the normal differentiation program, so that mature nondividing cells are not formed by the physiologic inducer of differentiation. However, even these tumors may still be induced to differentiate to form nonmalignant cells by treatment with compounds that can reverse the constitutive to the nonconstitutive state or induce the differentiation program by other pathways. In some tumors, such as sarcomas, reversal of malignancy can be obtained by specific changes in the karyotype [20, 61-63, 81]. But the stopping of cell division in mature cells by inducing differentiation induces a reversion of malignancy by bypassing the genetic changes that produce the malignant phenotype.
This research is now being supported by a contract with the National Foundation for Cancer Research, Bethesda, and by grants from the Jerome A. and Estelle R. Newman Assistance Fund, and the Julian Wallerstein Foundation.
1. Austin PE, McCulloch EA, Till JE (1971) Characterization of the factor in L cell conditioned medium capable of stimulating colony formation by mouse marrow cells in culture. J Cell Physiol77: 121-134 2. Azumi J, Sachs L (1977) Chromosome mapping of the genes that control differentiation and malignancy in myeloid leukemic cells. Proc Natl Acad Sci USA 74:253-257 3. Baccarani M, Tura S ( 1979) Correspondence, differentiation of myeloid leukemic cells: new possibilities for therapy. Br J Haematol 42:485-487 4. Bradley TR, Metcalf D ( 1966) The growth of mouse bone marrow in vitro. Aust J Exp BioI Med Sci 44: 287-300 5. Burgess A W, Metcalf D (1980) Characterisation of a serum factor stimulating the differentiation of myelomonocytic leukemic cells. Int J Cancer 26:647-654 6. Burgess AW, Camakaris J, MetcalfD (1977) Purification and properties of colony-stimulating factor from mouse lung conditioned medium. J BioI Chem 252: 1998-2003 7. Cohen L, Sachs L (1981) Constitutive gene expression in myeloid leukemia and cell competence for induction of differentiation by the steroid dexamethasone. Proc Natl Acad Sci USA 78: 353-357 8. Dewey MJ, Martin DW Jr, Martin GR, Mintz B (1977) Mosaic mice with terato carcinoma-derived mutant cells deficient in hypoxanthine phosphoribosyltransferase. Proc Natl Acad Sci USA 74: 5564-5568 9. Doolittle RF, Hunkapiller MW, Hood LE, Devare SG, Robbins KC, Aaronson SA ( 1983) Simian sarcoma virus onc gene, v-sis, is derived from the gene ( or genes) encoding a platelet-derived growth factor. Science 221:275-277 10. Falk A, Sachs L (1980) Clonal regulation of the induction of macrophage and granulocyte inducing proteins for normal and leukemic myeloid cells. Int J Cancer 26: 595 -601 II. Fibach E, Sachs L (1974) Control of normal differentiation of myeloid leukemic cells. IV. Induction of differentiation by serum from endotoxin treated mice. J Cell Physiol 83: 177-185 12. Fibach E, Sachs l (1975) Control of normal differentiation of myeloid leukemic cells. VIII. Induction of differentiation to mature granulocytes in mass culture. J Cell Physiol 86:221-230 13. Fibach E, Sachs L (1976) Control of normal differentiation of myeloid leukemic cells. XI. Induction of a specific requirement for cell viability and growth during the differentiation of myeloid leukemic cells. J Cell Physiol 89: 259-266 14. Fibach E, Landau T, Sachs L (1972) Normal differentiation of myeloid leukemic cells induced by a differentiation-inducing protein. Nature New BioI 237:276-278 15. Fibach E, Hayashi M, Sachs L (1973) Control of normal differentiation of myeloid leukemic cells to macrophages and granulocytes. Proc Natl Acad Sci USA 70: 343-346 16. Friend C (1978) The phenomenon of differentiation in murine erythroleukemic cells. Harvey Lectures 72. Academic, New York, pp 253-281 17. Ginsburg H, Sachs l (1963) Formation of pure suspension of mast cells in tissue culture by differentiation of lymphoid cells from the mouse thymus. J Natl Cancer Inst 31:1-40 18. Gootwine E, Webb CG, Sachs L (1982) Participation of myeloid leukaemic cells injected into embryos in haematopoietic differentiation in adult mice. Nature 299:63-65 19. Hayashi M, Fibach E, Sachs L (1974) Control of normal differentiation of myeloid leukemic cells. V. Normal differentiation to aneuploid leukemic cells and the chromo some banding pattern of D+ and D- clones. Int J Cancer 14:40-48 20. Hitosumachi S, Rabinowitz Z, Sachs l (1971) Chromosomal control of reversion in transformed cells. Nature 231 :511-514 21. Hoffman-liebermann B, Sachs L (1978) Regulation of actin and other proteins in the differentiation of myeloid leukemic cells. Cell 14: 825-834 22. Hoffman-Liebermann B, liebermann D, Sachs L (1981) Control mechanisms regulating gene expression during normal differentiation of myeloid leukemic cells. Differentiation defective mutants blocked in mRNA production and mRNA translation. Dev Bioi 81:255-265 23. Hoffman-liebermann B, Liebermann D, Sachs L (1981) Regulation of gene expression by tumor promoters. III. Complementation of the developmental program in myeloid leukemic cells by regulating mRNA production and mRNA translation. Int J Cancer 26:615-620 24. Housset M, Daniel MT, Degos L ( 1982) Small doses of Ara-C in the treatment of acute myeloid leukemia: differentiation of myeloid leukemia cells? Br J Haematol 51 : 125-129 25. Ichikawa Y (1969) Differentiation of a cell line of myeloid leukemia. J Cell Physiol 74: 223-234 26. Ichikawa Y, Pluznik DH, Sachs l (1966) In vitro control of the development of macrophage and granulocyte colonies. Proc Natl Acad Sci USA 56:488-495 27. Ichikawa Y, Pluznik DH, Sachs L (1967) Feedback inhibition of the development of macrophage and granulocyte colonies. I. Inhibition by macrophages. Proc Natl Acad Sci USA 58: 1480-1486 28. Ichikawa Y, Maeda N, Horiuchi M (1976) In vitro differentiation of Rauscher virus induced myeloid leukemic cells. Int J Cancer 17:789-797 29. Kasuga M, Fujita- Yamaguchi Y, Blithe Dl, Kahn CR (1983) Tyrosine-specific protein kinase activity is associated with the purified insulin receptor. Proc Natl Acad Sci USA 80: 2137-2141 30. Krystosek A, Sachs l (1976) Control oflysozyme induction in the differentiation of myeloid leukemic cells. Cell 9: 675-684 31. Landa uT, Sachs L ( 1971) Characterization of the inducer required for the development of macrophage and granulocyte colonies. Proc Natl Acad Sci USA 68:2540-2544 32. Liebermann D, Hoffmann-liebermann B, Sachs l ( 1980) Molecular dissection of dif ferentiation in normal and leukemic myelo blasts: separately programmed pathways of gene expression. Dev Bioi 79:46-63 33. Liebermann D, Hoffman-Liebermann B, Sachs L (1982) Regulation and role of different macrophage and granulocyte proteins in normal and leukemic myeloid cells. Int J Cancer 29: 159-161 34. Lipton J, Sachs L ( 1981) Characterization of macrophage and granulocyte inducing proteins for normal and leukemic myeloid cells produced by the Krebs ascites tumor. Biochim Biophys Acta 673: 552-569 35. Lotem J, Sachs L (1974) Different blocks in the differentiation of myeloid leukemic cells. Proc Natl Acad Sci USA 71: 3507 -3511 36. Lotem J, Sachs L (1975) Induction of specific changes in the surface mem brane of myeloid leukemic cells by steroid hormones. Int J Cancer 15: 731- 740 37. Lotem J, Sachs L (1977a) Control of normal differentiation of myeloid leukemic cells. XII. Isolation of normal myeloid colonyforming cells from bone marrow and the sequence of differentiation to mature granulocytes in normal and D+ myeloid leukemic cells. J Cell Physiol92: 97-108 38. Lotem J, Sachs L ( 1977 b) Genetic dissection of the control of normal differentiation in myeloid leukemic cells. Proc Natl Acad Sci USA74:5554-5558 39. Lotem J, Sachs L (1978a) Genetic dissociation of different cellular effects of interferon on myeloid leukemic cells. Int J Cancer 22:214-220 40. Lotem J, Sachs L (1978b) In viva induction of normal differentiation in myeloid leukemic cells. Proc Natl Acad Sci USA 75:3781 -3785 41. Lotem J, Sachs L (1979) Regulation of normal differentiation in mouse and human myeloid leukemic cells by phorbol esters and the mechanism of tumor promotion. Proc Natl Acad Sci USA 76: 5158-5162 42. Lotem J, Sachs L (1980) Potential prescreening for therapeutic agents that induce differentiation in human myeloid leukemic cells. Int J Cancer 25:561-564 43. Lotem J, Sachs L (1981) In viva inhibition of the development of myeloid leukemia by injection of macrophage and granulocyte inducing protein. Int J Cancer 28: 375-386 44. Lotem J, Sachs L (1982) Mechanisms that uncouple growth and differentiation in myeloid leukemia: restoration of requirement for normal growth-inducing protein without restoring induction of differentiation-inducing protein. Proc Natl Acad Sci USA79:4347-4351 45. Lotem J, Sachs L (1983a) Coupling of growth and differentiation in normal myeloid pre cursors and the breakdown of this coupling in leukemia. Int J Cancer 32: 127 -134 46. Lotem J, Sachs L (1983b) Control ofin viva differentiation of myeloid leukemic cells. III. Regulation by T lymphocytes and inflammation. Int J Cancer 32: 781- 791 47. Lotem J, Sachs L (1984) Control of in viva differentiation of myeloid leukemic cells. IV. Inhibition of leukemia development by myeloid differentiation-inducing protein. Int J Cancer 33: 147-154 48. Lotem J, Lipton J, Sachs L (1980) Separation of different molecular forms of macrophage and granulocyte inducing proteins for normal and leukemic myeloid cells. Int J Cancer 25:763-771 49. Maeda M, Horiuchi M, Numa S, Ichikawa y ( 1977) Characteriza tion of a differen tia tion stimulating factor for mouse myeloid leukemic cells. Gann 68: 435-447 50. Marks P, Rifkind RA (1978) Erythroleukemic differentiation. Ann Rev Biochem 47: 419-448 51. Metcalf D ( 1969) Studies on colony formation in vitro by mouse bone marrow cells. I. Continuous cluster formation and relation of clusters to colonies. J Cell Physiol 74: 323 -332 52. Michalewicz R, Lotem J, Sachs L (1984) Cell differentiation and therapeutic effect of low doses of cytosine arbinoside in human myeloid leukemia. Leuk Res (in press) 53. Mintz B, Illmensee K (1975) Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci USA72:3585-3589 54. Moore MAS (1982) G-SCF: its relationship to leukemia differentiation-inducing activity and other hemopoietic regulators. J Cell Physiol [Suppl] 1: 53-64 55. Nicola NA, Burgess AW, Metcalf D (1979) Similar molecular properties of granulocytemacrophage colony stimulating factors produced by different organs. J BioI Chem 24:5290-5299 56. Paran M, Ichikawa Y, Sachs L (1968) Production of the inducer for macrophage and granulocyte colonies by leukemic cells. J Cell PhysioI72:251-254 57. Paran M, Sachs L, Barak Y, Resnitzky P (1970) In vitra induction of granulocyte differentiation in hematopoietic cells from leukemic and non-leukemic patients. Proc Natl Acad Sci USA 67: 1542-1549 58. Pike B, Robinson WA (1970) Human bone marrow growth in agar gel. J Cell Physiol 76:77-84 59. Pluznik DH, Sachs L (1965) The cloning of normal "mast" cells in tissue culture. J Cell Comp Physio166:319-324 60. Pluznik DH, Sachs L (1966) The induction of clones of normal "mast" cells by a substance from conditioned medium. Exp Cell Res 43:553 -563 61. Rabinowitz Z, Sachs L ( 1968) Reversion of properties in cells transformed by polyoma virus. Nature 220: 1203-1206 62. Rabinowitz Z, Sachs L (1970) Control of the reversion of properties in transformed cells. Nature 225: 136-139 63. Sachs L (1974) Regulation of membrane changes, differentiation, and malignancy in carcinogenesis. Harvey Lectures 68. Academic, New York, pp 1-35 64. Sachs L (1978a) Control of normal cell differentiation and the phenotypic reversion of malignancy in myeloid leukemia. Nature 274:535-539 65. Sachs L (1978b) The differentiation of myeloid leukemia cells. New possibilities for therapy. Br J HaematoI40:509-517 66. Sachs L (1980) Constitutive uncoupling of pathways of gene expression that control growth and differentiation in myeloid leukemia: a model for the origin and progression of malignancy. Proc Natl Acad Sci USA 77:6152-6156 67. Sachs L (1982a) Control of growth and differentiation in leukemic cells: regulation of the developmental program and restoration of the normal phenotype in myeloid leukemia. J Cell Physiol [Suppl] 1: 151-164 68. Sachs L (1982b) Normal developmental programmes in myeloid leukemia: regulatory proteins in the control of growth and differentiation. Cancer Surveys 1: 321-342 69. Simantov R, Sachs L (1978) Differential desensitization of functional adrenergic receptors in normal and malignant myeloid cells. Relationship to receptor mediated hormone cytoxicity. Proc Natl Acad Sci USA 75:1805-1809 70. Simantov R, Shkolnik T, Sachs L ( 1980) Desensitization of enucleated cells to hormones and the role of cytoskeleton in control of a normal hormonal response. Proc Natl Acad Sci USA 77:4798-4802 71. Stanley ER, Heard PM (1977) Factors regulating macrophage production and growth. Purification and some properties of the colony stimulating factor from medium conditioned by mouse L cells. J Biol Chem 252: 4305-4312 72. Symonds G, Sachs L (1982a) Autoinduction of differentiation in myeloid leukemic cells: restoration of normal coupling between growth and differentiation in leukemic cells that constitutively produce their own growth-inducing protein. EMBO J I: 1343 -1346 73. Symonds G, Sachs L (1982b) Cell competence for induction of differentiation by insulin and other compounds in myeloid leukemic clones contin uously cultured in serum-free medium. Blood 60: 208-212 74. Symonds G, Sachs L (1982c) Modulation of cell competence for induction of differentiation in myeloid leukemic cells. J Cell Physiollll : 9-14 75. Symonds G, Sachs L (1983) Synchrony of gene expression and the differentiation of myeloid leukemic cells: reversion from constitutive to inducible protein synthesis. EMBO J 2:663-667 76. Ushiro H, Cohen S ( 1980) Identification of phosphotyrosine as a product of epidermal growth factor-activated protein kinase in A-431 cell membranes. J BioI Chem 225 : 8363-8365 77. Waterfield MD, Scrace GT, Whittle N, Stroobant P, Johnsson A, Wasteson A, Westermark B, Heldin CH, Huang JS, Oeuel TF (1983) Platelet-derived growth factor is structurally related to the putative transforming protein p28sIs of simian sarcoma virus. Nature 304: 35-39 78. Webb CG, Gootwine E, Sachs L (1984) Oevelopmental potential of myeloid leukemia cells injected into mid-gestation embryos. Dev Biol101:221-224 79. Weisinger G, Sachs L (1983) DNA-binding protein that induces cell differentiation. EMBO J 2:2103-2107 80. Weiss B, Sachs L (1978) Indirect induction of differentiation in myeloid leukemic cells by lipid A. Proc Natl Acad Sci USA 75: 1374 -1378 81. Yamamoto T, Rabinowitz Z, Sachs L (1973) Identification of the chromosomes that control malignancy. Nature (New BioI) 243:247 -250 82. Yamamoto Y, Tomida M, Hozumi M (1980) Production by spleen cells of factors stimulating differentiation of mouse myeloid leu kemic cells that differ from colony stimulating-factor. Cancer Res 40:4804-4809 |