|
1 Dept. of Molecular Biology Bionetics Research
Foundation 7300 Pearl St. Bethesda, Md. 20014
Normal function of hemopoietic tissues depends on a continuous supply of functional mature cells through maturation of progenitor cells and stem cells. Leukemia is believed to be resulted from a disturbance in this maturation process. In mouse, the stem cells are measured by spleen colony technique (CFU-S measurement) and the progenitor cells by in vitro culture technique (CFU-C measurement). Sachs and his colleagues reported that human leukemic cells can be induced to mature in the in vitro culture system in the presence of colony-stimulating activity (CSA). These findings were partially confirmed and extented to many leukemic cases. Our attempt to establish a relationship between the type of leukemia and nature of leukemic cell maturation in culture was not successful. Using RL V induced leukemia in mice as a model system, the effect of RL V infection on the number of CFU-C and CFU-S was studied. Our results showed that there was an early transitory depression of CFU-S and a long-term stimulation of CFU-S following RL V infection. These results suggest that the earliest event following RL V infection is the committment of CFU-S to CFU-C and therefore CFU-S are target cells of RL v. However, these conclusions do not exclude the possibility that the other cell types could also be target cells. The mature granulocytes derived from leukemic cells show budding virus particles. The budding virus particles were also found in mature granulocytes and anucleated erythrocytes obtained from RL V infected mice.
The normal function of hemopoiesis is carried out by various kinds
of mature blood cells, such as erythrocytes, granulocytes) megakaryocytes
and lymphoid cells. The life span of these mature cells are relatively
short. Therefore, a continuous supply of these mature cells is essential
for the normal function of the hemopoietic tissue. These mature
cells are derived from a class of early undifferentiated cells through
a sequence of differentiation and proliferation. This class of early
undifferentiated cells are termed "stem cells" (1, 2). It is important
to emphasize that the stem cells are defined according to their
functions. The morphology of this class of cell is not known. A
stem cell should have the following properties. It should be capable
of I) proliferation, 2) differentiation, 3) self-renewal upon proliferation
and 4) response to physiological demands. If a stem cell is capable
of giving rise to more than one type of differentiated cells, it
is termed a multipotent stem cell whereas if it is capable of giving
rise only to one type of differentiated cell it is termed unipotent
stem cell. A multipotent stem cell by definition is a precursor
of a unipotent stem cell (progenitor cell). Their relationship is
diagrammatically illustrated in Fig. 1. This figure depicts the
transition from stem cell to progenitor cell and the transition
from progenitor cell to differentiated cell which are dependent
on the presence of specific factors. The production of these regulatory
factors presumably are under physiological control. Any cause which
disturbs the process of maturation shown in Fig. 1 will naturally
impair the normal function of hemopoietic tissue. Leukemia is believed
to result from a disturbance of this maturation process ( 3-8) .There
are three possible outcomes that could result from this disturbance
( see Fig. 2) .These are: ( A ) blockage of differentiation ( 4-8)
, ( B) aberration of differentiation and (C) reversal of differentiation.
In (A) and (C) , the leukemic cells should represent some cell types
present during the course of normal differentiation and in ( B )
, the leukemic cells should be different from any of the known cell
types. The mnodels in this figure also indicate that the target
cells of leukemogenesis (with respect to their stage of differentiation)
are not well defined. So far there is no conclusive evidence to
support or rule out any of these possibilities or to implicate a
particular stage of cell differentiation as the target cell for
the initiation of leukemogenesis. 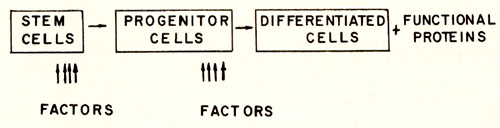
II. Measurement of Stem Cells Currently there are two functional assays available, one for stem cells, the other for progenitor cells. Both of them are based on clonal methods. They are summarized in Table 1. In mice, stem cells can be assayed by the spleen colony technique ( 2) .The assay is performed by injecting an adequate number of nucleated cells into a lethally irradiated recipient. After a period of 8-10 days, the spleen is removed. The macro-nodules which develop on the surface of the spleen are then counted. These nodules are called spleen colonies. Each colony was shown to derive from a multipotent stem cell as judged by a chromosomal analysis using radiation induced chromosomal aberrations as markers (9) and by studying the cellular composition of each colony (10,11). Apparently, the development of a spleen colony represents a response of a stem cell to a physiological demand after irradiation and is regulated by the microenvironment (12) which contain cells that produce short-term mediated h umo ral factors ( 13) . The progenitor cell for granulocytic cells can be assayed by an in vitro method originally developed by Bradley and Metacalf (14) and by Pluznik and Sachs (15). A single cell suspension is prepared in a semi-soft medium ( either 0.3% agar [14, 15] or 0.8% methyl cellulose [16]) in the presence of colony stimulating activity (CSA). The CSA is supplied either in the same layer as the cell suspension ( 16) or in a separate layer of 0.5 % agar containing conditioned medium or feeder cells ( 14, 15) .The conditioned medium was generally prepared by growing cell population containing factor producing cells in culture medium ( either in the presence or in the absence of Table 1. Colony Methods for the Assay
of Haemopoietic Stem Cells and Progenitor Cells  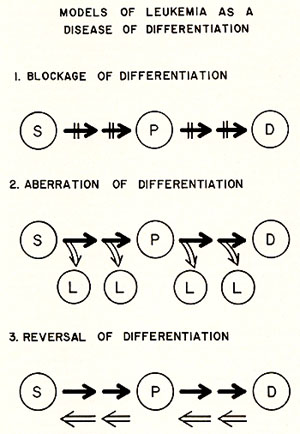
fetal calf serum) for 4 to 6 days ( 15) .The number of colonies is generally recorded 8 to 10 days after plating. At this stage, the colony size may be as large as 104 cells. The colonies are classified as granulocytic or macrophage colonies depending on the composition of cells in the colonies. Both of these colony types contain cells at various stages of maturation. Iferythropoietin is present in this culture system the second day after plating, some very small colonies containing erythropoietic cells ( 8 --100 cells ) may develop ( 17, 18) .In mice, colony formation is strictly dependent on the addition of CSA. CSA for assaying mouse colony forming cells can be obtained from mouse sera (19), human urine (20), medium from cultured fibroblasts (21 ), and from embryonic primary culture cells (22). CSA are glycoproteins which can be purified by use of concanavalin Asepharose chromatography. The molecular weight estimates of this activity have indicated heterogeneity ranging from 10,000 to 190,000 (23 ). It is not known whether CSA is a population of heterogenous glycoproteins or a complex protein containing many subunits. This in vitro culture assay for progenitor cells has been successfully applied to human blood cells ( 24-27) .Similar to colonies derived from mouse cells, the colonies derived from human blood cells (CFU-C) also contain granulocytic cells at various stages of maturation, but in general, the colony sizes of human origin are much smaller ( 50-1,000 cells) than those of mouse origin. The specificity of CSA required for the in vitro colonies of human origin also differs from that of mouse origin. In most cases, CSA prepared from human cells can stimulate the growth of colonies of both human and murine origin, but the CSA prepared from mouse cells stimulates only mouse colonies. One difficulty in obtaining a quantitative measurement of human colony forming cells is the lack of an absolute requirement for exogenous CSA for colony formation. This is due to a contamination of factor-producing cells in the human blood cell population. A successful attempt to separate the factor producing cells from the colony forming cells has recently been reported (28). Obviously, separation of these two cell populations is essential for a successful study of the maturation process.
One of the most important observations made during the course of studies of human progenitor cells in culture is that apparently some leukemic blast cells can be induced to mature in culture in the presence of appropriate CSA to apparently mature normal appearing granulocytes (25, 29). The evidence that these mature cells are derived from leukemic cells is based on some chromosomal analyses (30,31 ). But the argument is not conclusive, because one still cannot rule out the possibility that the mature cells are derived from normal cells which contains the same chromosomal aberration. However, since the disclosure of maturation of human leukemic cells in culture, many studies have been carried out to establish a pattern as to which type of leukemic cells seems to be "inducible" to mature. The result of these studies are also inconclusive. A summary of such studies carried out in our laboratory in the past year is shown in Table 2. The capacity of colony formation varies with various types of leukemia and also varies within the same type of leukemia. This variation might be due to either difficulty in assuring reproducibly standardized in vitro colonies assay Table 2. Differentiation of Leukemic
Cells in Culture 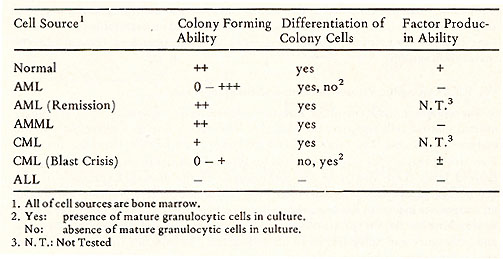
The apparent inducement of human leukemic cells to develop into
seemingly normal mature cells supports the idea that leukemia is
due to a disturbance in the maturation process. The cause of this
disturbance is not known. However, there are now strong reasons
for suspecting a role for type-C RNA tumor viruses or information
derived from these viruses in the pathogenesis of the disease. Expression
of viral genomes in human leukemic cells either from endogenous
genetic information or from an exogenous infection has been recently
supported from results of extensive studies in searching for the
footprints of viral information in human leukemic cells by Gallo
and colleagues and Spiegelman and his colleagues. The evidence for
the existence of viral information in human cells is as follows:
1) Virus-like reverse transcriptases have been isolated from some
human leukemic cells but not from normal proliferative cells. These
enzymes have the biochemical characteristics like most of known
mammalian viral reverse transcriptases (33-35). For example, this
enzyme activity is sensitive to RNAse. The enzymes are able to transcribe
the heterogenous part of 70S RNA isolated from primate and mammalian
viruses. They prefer ( dT) 12 -18. (rA)n as template-primer over
( dT) 12 -18. ( dA)n and are able to use ( dG ) 12 -18. ( rC)n.
These are the characteristics of viral reverse transcriptase ( 35,
36) ; 2) The leukemic reverse transcriptase is immunologically related
to reverse transcriptase of primate type-C RNA tumor viruses (37,
38). (They were inhibited by antibodies against viral reverse transcriptase
expecially of primate, but also of murine RNA tumor viruses [37,38].
This suggests that the human leukemic enzyme shares a common antigenic
determinant with primate and murine viral enzymes. ) ; 3) Human
leukemic cells contain an RNA sequence homologous to DNA products
prepared from an endogenous reaction of mammalian and primate RNA
oncogenic viruses (39,40,41); 4) The size of the RNA template-primer
for the leukemic reverse transcriptase appears to be the same as
viral RNA ( 42, 43) ; 5) The reverse transcriptase and associated
RNA are present in a post-mitochondrial cytoplasmic particle which
retains morphological integrity on repeated high speed centrifugation
and has a density of 1.15 to 1.18 ( 43) , characteristics typical
of type-C RNA tumor viruses. It now appears unequivocal that these
particles are viral-related. This information has been recently
reviewed in detail elsewhere ( 44 ). V. RNA Oncogenic Viruses and Murine Stem Cells With the above background in mind, it is evident that studies designed
to determine the affect of animal type-C viruses on normal hemopoiesis
are of immediate interest. The data described below was derived
from our beginning attempts in this direction in mice. One advantage
of an animal system is that one can follow a defined course of leukemic
development, especially the early events following viral infection.
Informa- 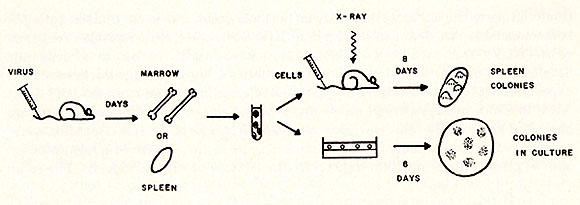 Fig. 3: Experimental design for the effect of RL V infection on cru-s and cru-c compartments. Eight-week old NIH Swiss mice were used for this experiment. RL V was injected intravenously through the tail vein. To prepare cell suspensions, at least five uninfected and infected mice were used, respectively, Spleen colony technique was performed as described previously (2). The cru-c was measured according to the procedures described by Pluznik and Sachs ( 15) , except that CSA was prepared from primary culture of BALB/C embryonic cells. 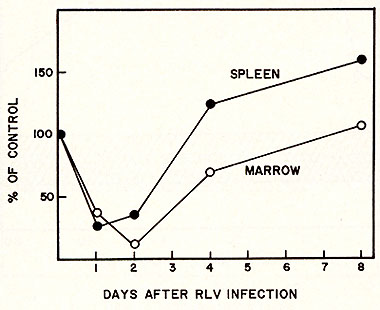
tion relating to the earliest events may be the most important
to our understanding of leukemogenesis. We induced leukemia in NIH
Swiss mice with Rauscher leukemia virus (RL V) and used it as a
model system to study the effect of leukemia development on the
process of leukocyte maturation. The experimental design of this
experiment is shown in Figure 3. RL V was injected intravenously
into NIH Swiss mice, and various days after injection, marrow and
spleen cells of infected mice were harvested and a single-cell-suspension
was made. Aliquots of the cell suspension were: ( a) injected into
the lethally irradiated mice to measure the content of spleen colonies,
and (b) plated on agar medium to measure the content of progenitor
cells. The result 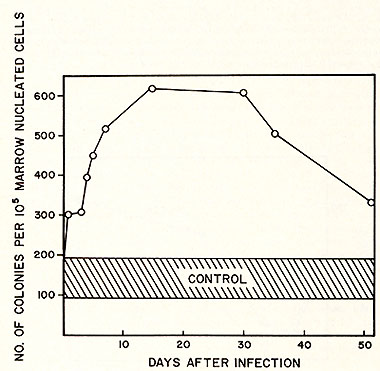
of this experiment for spleen colony formation is shown in Figure
4. There is an initial dip in the number of colony forming units
(CFU-S) per unit cell number injected both from spleen and marrow
cells on the second and third days after injection of RL v. However,
the CFU-S per unit cell number then gradually recovered to a normal
level. A short period of "overshoot" was observed on the fourth
and fifth day after RL V injection. Since the cell number increased
during the course of leukemia development, the total CFU-S, in fact,
increased more than 10 fold by one month following the development
of leukemia. The effect of RL V infection on the progenitor compartment
of marrow is shown in Figure 5. Soon after viral infection, the
number of CFU-C (granulocytic progenitor cells) per 105 nucleated
cells increased about 10 fold. This level was maintained for a few
weeks and then gradually decreased by the fifth week after infection.
We did not determine whether CFU-C eventually decreases to a normal
or a subnormal level since infected animals started to die by the
eighth week after inoculation with RL V. This rise and decline of
CFU-C following leukemic development might provide an explanation
for the variation in measuring CFU-C in human leukemic cell populations.
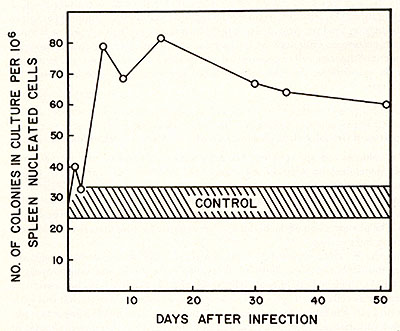
The number of CFU-C may be a function of the stage of leukemic development. Similar results were obtained when spleen cells were used to study the effect of CFU-C upon viral infection. The result of this experiment is shown in Figure 6. This graph was plotted as CFU-C per unit cell number as a function of time. Since spleen cell number increased at least 20 fold during the development of leukemia, the total CFU-C increased at least 120 fold. From a practical point of view, we wonder whether this increase of CFU-C in early infection might be useful as a diagnostic tool for pre-leukemia or an early stage of leukemia. The effect on the number of CFU-C and CFU-S by RL V were not simply due to an antigenic stimulation since heat-inactivated viruses did not cause such an effect. Some agents such as Freund's complete adjuvant (45), pertussis vaccine (46), phenylhydrazine ( 47 ) , and endotoxin ( 48) are able to enhance the number of CFU-C and CFU-S in vivo. However, these effects are transitory and, therefore, are different from the long-term effect of RL V. Our observation on the initial decrease of CFU-S by RL V infection is in agreement with that reported by Seidel ( 49) as is our finding on the late effect of CFU-S by RL V infection ( 49 ) and this late effect is also similar to that reported by Okunewick, et at. (50). Many interpretations can account for the initial decrease of CFU-S accompanying the increase of CFU-C. One interpretation is that the CFU-S is a target cell of RL V. Upon infection, the stem cells are affected such that their capabilities for spleen colony formation are lost. Now, these infected stem cells are still capable of forming colonies in culture at evidenced by an increase of CFU-C. Perhaps this occurs by a commitment of the stem cell to granulocytic progenitors. However, our results did not rule out the possibilities that the other cell compartment could also be a target for RL V infection.
1) The colonies contain apparent mature granulocytes and colony
formation is absolutely dependent on addition of exogenous CSA.
We found type-C RNA tumor viruses bud from the membrane of the mature
granulocytes. In addition, on examining the other cell types from
infected mice, we found that besides early immature cells, both
mature granulocytes and anucleated erythrocytes show budding type
virus particles. The phenomenon of the budding of virus particles
from mature erythrocytes is interesting and puzzling.
Leukemia appears to result from a disturbance of the normal differentiation of hemopoietic stem cells. This disturbance might be a blockage, aberration or reversal of differentiation. In some cases, the blockage process of some cells can apparently be removed in culture as evidenced by apparent induction of leukemic cells to produce seemingly normal mature granulocytes in culture in the presence of CSA. This dependence of CSA on the maturation of leukocytes may cast some light on the process of leukemogenesis. The phenomenon of CSA stimulation may provide avenues for studying the process of leukocyte maturation at the molecular level and a potential new approach for the therapy of human leukemia and other blood illnesses. In this respect, however, it is surprising and puzzling that so far there are no reports evaluating in vivo results of treatment of leukemic mice with CSA.
1. McCulloch, E. A. (1966) In, "The Physiological Basis of Medical Practice." 8th ed., Chapter 32, p. 508, C. H. Best and N. B.Taylor (Eds.),WilliamsandWilkins, Baltimore. 2. Till, J. E. and McCulloch, E. A. ( 1961) Rad. Res., 14: 213. 3. McCulloch,E.A.andTill,J.E. (1971)Amer.I.Path., 65: 737. 4. Ginsburg, H. and Sachs, L. (1965) I. Cell Comp. Physiol., 66: 199. 5. Sachs, L. (1970) In, Regulation ofHematopoiesis, Appleton,.Century-Crofts, New York,Vol. I,p. 217. 6. Gallo,R.C.(1971)ActaHaemat.,45: 136. 7. Perry, S. and Gallo, R. C. (1970) In, Regulation ofHemopoiesis, A. Gordon (Ed.) Appleton-Century-Crofts, New York, P. 1221. 8. Gallo, R. C. (1973) On the Etiology of Human Acute Leukemia, Med. Clin. of North America, 57: 343. 9. Becker,A.J.,McCulloch,E.A.,andTill,J.E.(1963)Nature, 197:452. 10. Fowler, J. H., Wu, A. M., Till, u. D., Siminovitch, L., and McCulloch, E. A. (1967) ]. Cell Physiol., 69: 65. 11. Wu, A. M., Till, ]. E., Siminovitch, L., and McCulloch, E. A., (1967) ]. Cell Phys.iol., 69: 177. 12. Curry,J. L. and Trentin,].]. (1967)Develop. BioI., 15: 395. 13. McCulloch, E. A., Gregory, C. J., and Till, ]. E. (1973) Symp. for Ciba Foundation, in press. 14. Bradley, J.R. and Metcalf, D. (1966) Aust. J. Expt. BioI. Med. Sci., 44: 287. 15. Pluznik, D. H. and Sachs, L. (1965)]. Cell Comp. Physiol., 66: 319. 16. Worton, R. G., McCulloch, E. A., and Till, ]. E. (1969)]. Cell Physiol., 76: 171. 17. Stephenson,]. R., Axelrod, A. A., McLead, D. L., and Shreeve, M. M. (1971) Proc. Nat. Acad. Sci., USA, 69: 1542. 18. Isocove, N. (1973) In, The Second International Workshop on Hemopoiesis in Culture, sponsored by N. C. I. Medical Oncology Area, Virginia, Proc. Nat. Acad. Sci. , in press. 19. Stanley, E. R., Robinson, W., Ada, G. L. (1968) Aust. . Expt. BioI. Med. Sci., 46 : 715. 20. Robinson, W. A., Stanley, E. R., and Metcalf, D. (1969) Blood, 33: 396. 21. Landau, T. and Sachs, L. (1971)Proc. Nat. Acad. Sci., USA, 68: 2540. 22. Paran, M. and Sachs, L. (1969)]. Cell Physiol., 73: 91. 23. Metcalf, D. and Moore, M. A. S. (1971) In, Hemopoietic Cells, Chapter 7, p. 403. A. Neuberger and E. L. Tatum (Eds.), North Holland Publishing Company, Amsterdam -London. 24. Senn,J.G.,McCulloch,E.A.,andTill,].E.(1967)Lancet,2: 597. 25. Paran, M., Sachs, L., Barak, T ., and Resinitzky, P. ( 1970) Proc. Nat. Acad. Sci., USA, 67: 1542. 26. Pike, B. L. and Robinson, W. A. (1970 )]. Cell Physiol., 76: 77. 27. Brown, III, C. H. and Carbone, P. (1971)]. Nat'l. Cancer Inst., 46: 989. 28. Messner, H. A., Till,] .E., and McCulloch, E. A. (1973) Blood, in press. 29. Moore, M. A. S., Williams, N., and Metcalf, D. (1973)]. Nat. Cancer Inst., 50: 603. 30. Moore, M. A. S. and Metcalf, D. (1973) Int. ]. Cancer (submitted for publication). 31. Duttera,M.].,Whang-Peng,].,andBull,].M.(1972)Lancet, 1: 715. 32. Robinson, W. A. (1973) In, The Second Int. Workshop on Hemopoiesis in Culture, sponsored by N. C. I. Medical Oncology Area, Virginia, in press. 33. Gallo, R. C., Yang, S. S., and Ting, R. C. (1970) Nature, 228: 972. 34. Sarngadharan, M. G., Sarin, P. S., Reitz, M. S., and Gallo, R. C. (1972) Nature New Biology, 240: 67. 35. Gallo, R. C., Sarin, P. S., Smith, G., Bobrow, S. N., Sarngadharan, M. G., Reitz, M. S., Abrell, ]. W ., In, Proceedings of the 2nd Annual Steenbock Symposium, DNA Synthesis in Vitro, in press. 36. Gallo, R. C., Sarin, P. S., Sarngadharan, M. G., Smith, R. G., Bobrow, S. N., and Reitz, M. S., In, Proceedings of the Sixth Miles International Symposium on Molecular Biology, Beers, P. F. ( Ed. ) , in press. 37. Todaro, G. ]. and Gallo, R. C. (1972) In, The IV Lepetit Colloquium "Possible Episomes in Eukaryocytes", L. Silvestri (Ed.), North Holland Publishing Co., Amsterdam-London, in press. 38. Todaro,C.J.and Gallo,R.C.,(1973)Nature, 244: 206. 39. Baxt, W ., Hehlmann, R., and Spiegelman, S. (1972) Nature New Bioi., 240: 72. 40. Baxt, W. and Spiegelman, S. (1972) Proc. NNat. Acad. Sci., USA, 69: 3737. 41. Callo, R. C., Miller, N., Saxinger, W. C., and Cillespie, D., (1973) Proc. Nat. Acad. Sci., USA, 70: 3219. 42. Axel, R., Schlom, J ., and Spiegelman, S. ( 1972) Nature, 235: 32. 43. Callagher, R. C. and Callo, R. C. (1973) This Symposium. 44. Sarin, P. and Callo, R. C. (1973) In, International Review of Science, Series in Biochemistry, Vol. 6, Nucleic Acids, K. Burton (Ed.), Butterworth Medical and Technical Publishing Co., Oxford, Chap. 8. 45. Haskill, J. S., McNeill, T. A., and Moore, M. A. S. ( 1970) J. CellPhysiol., 75: 167. 46. Bradley, T. R., Metcald, D., Sumner, M., and Stanley, E. R. (1967) In, Characteristics of In Vitro Colony Formation By Cells From Hemopoietic Tissues, P. Barnes (Ed.), Williams and Wilkins Co., Baltimore, p. 22. 47. Stohlmann, F. (1973) personnal communication. 48. McNeill, T. A. (1970) Immunology, 18: 61. 49. Seidel, H. J. (1973) Z. Krebsforsch, 79: 123. 50. Okunewick, J. P ., Phillips. E. L., and Erhard, P. (1972) J. Nat. Cancer Inst., 49 : 1101. 51. Becker, A. J ., McCulloch, E. A., Siminovitch, L., and Till, J. E. (1965) Blood, 26 : 296. 52. Iscove, N., Till, J. E., andMcCulloch, E. A. (1970)Proc. Sco. Exp. Bioi. andMed., 134: 33. 53. Dixon, C. L., Dulmadge, E. A., ans Schabel, Jr., F. M. (1966) Cancer chemo therapy Rept., 50: 247. 54. Ichikawa,Y. (1969)J. CellPhysiol., 74: 223. |