|
Institute of Cancer Research, College of Physicians
& Surgeons, Columbia University, 99 Fort Washington Avenue, NewYork,N.Y.10032
I. The Strategy of the Search for RNA Tumor Viruses in Human
Malignancies
Our overall purpose has been and remains to explore the possible
involvement of RNA tumor viruses as etiologic or cofactor agents
in human neoplasias and to exploit any leads that emerge that could
be of any conceivable use in the prevention, diagnosis, or therapy
of human cancer . The task of identifying the existence and the
causative role of the animal RNA tumor viruses was inadvertently
made easier by breeding high cancer incidence animal strains. In
the process, a homogeneous genetic background was created that was
permissive for viral replication. As a consequence, virus particles
reached levels that made their detection inevitable. Those who are
concerned with human neoplasias are for the most part faced with
the same difficulties encountered by the 1 early animal oncologists
prior to the availability of inbred strains. It follows that a search
for putative human viral agents requires more sensitive devices
than those which sufficed to establish their presence in the genetically
homogeneous animal systems. In the quest for such tools, we quite
naturally turned to molecular hybridization and the other methodologies
developed by molecular biologists in the past several decades. Our
investigations evolved through a number of stages that are conveniently
identified by the questions we posed for experimental resolution
: 1) Do human neoplasias contain RNA molecules possessing detectable
homology to the RNA of tumor viruses known to cause similar cancers
in other mammals? 2) If a positive outcome is obtained, do the RNA
molecules identified in tumors possess the size and physical association
with reverse transcriptase that characterize the RN A of the animal
oncornaviruses ? 3) If such RNA exists in human tumors, is it encapsulated
in a particle possessing the density and size of the RNA tumor viruses?
4) Is the RNA of human tumor particles homologous to the RNA of
the viruses causing the corresponding disease in animals? 5) The
"virogene-oncogene" concept proposes that all animals prone to cancer
carry in their germ line a complete copy of the information required
to convert a cell from normal to malignant for the production of
tumor virus particles. Is this concept valid for randomly bred populations
and, in particular, for the human disease ?
II. The Animal Models as a Point of Departure
When we began our investigations, there were relatively few animal
oncornaviruses a vailable in amounts adequate for the sort of biochemical
experiments required. Table I lists these and records certain relevant
features that served as a
Table I: Comparsion of Some Representative Oncornaviruses
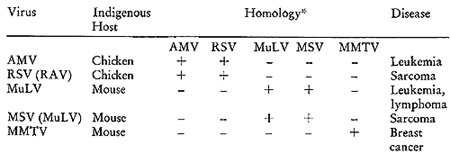
*The results of molecular hybridizations between [³H]DNA
complementary to the various RNAs and the indicated RNAs. The plus
sign indicates the hybridizations were positive and the negative
sign indicates none could be detected.
guide in these expermients. There are two avian viruses, myeloblastosis
virus (AMV) and the Rous sarcoma virus (RSV), that cause mesenchymal
malignancies in chickens. In addition, we have the murine leukemia
virus (MuL V) and the murine sarcoma virus (MuSV) that induce similar
diseases in mice. Finally, we have the murine mammary tumor virus
(MMTV), which is the unique etiologic agent for mammary tumors.
When these viruses are examined for sequence homologies amongst
their nucleic acids, a rather informative pattern emerges. I twill
be noted that the two chicken agents have sequences in common, but
do not show detectable homology with any of the murine agents. Turning
to the murine viruses, we find that the nucleic acids of the leukemia,
lymphoma, and sarcoma agents are homologous to one another, but
not to either of the two avian agents or to the mammary tumor virus.
Finally, the mouse mammary tumor virus has a singular sequence homologous
on]y to itself.
It is important to understand that a plus sign does not indicate
identity, but simply sufficient similarity to be detectable by the
relaxed hybridization conditions used in these initial studies.
Similarly, a negative sign does not imply the complete absence of
sequence homology, but rather that none was observable by the procedures
used.
If analogous, or similar, virus particles are associated with the
corresponding human diseases, certain predictions may be hazarded
on the basis of the specificity patterns exhibited in Table I, and
they may be listed as follows:
(a) In view of the lack of homology between the avian and murine
agents, it is unlikely, from simple evolutionary considerations,
that human agents, should they exist, would show more homology to
the avian group than to the murine oncornaviruses.
(b) It follows that the murine tumor viruses would represent the
more hopeful source of the molecular probes required to search for
similar information in the analogous human cancers.
(c) If particles are found to be associated with human mesenchymal
tumors (in leukemias, sarcomas, and lymphomas), their RNAs might
show homology to one another and possibly to that of the murine
leukemia virus.
(d) If RNA particles are identified in human breast cancer, they
should not exhibit homology to the RNA of virus-like particles associated
with the human mesenchymal neoplasias or to MuL V RNA, but might
exhibit some homology to the RNA of the murine mammary tumor virus.
On the basis of both availability and the specificity considerations
outlined above, it is clear why the murine agents were initially
chosen for producing the necessary molecular probes to look for
corresponding information in the human disease. Furthermore, the
desire to monitor the biological consistency of our findings dictated
that we examine in parallel the human neoplasias listed. Such a
parallel examination would permit us to determine whether our findings
in humans mirrored biologically what was known from the animal experimental
models. For this purpose, we focused our efforts on the mesenchymal
neoplasias and on breast cancer.
III. Molecular Hybridization with Radioactive DNA Probes
The DNA-RNA hybridization procedure we used to answer the question
whether human tumors contain viral-related RNAs was one that we
had designed (1) some fifteen years ago to answer questions of almost
precisely this nature in the case of virus-infected bacteria. The
method depends on the ability of any piece of single-stranded DNA
to find its complementary RNA and form, under the proper conditions,
a double-stranded hybrid structure. The reaction is highly specific
and has proved to be of considerable value in molecular biology
over the past decade.
The required radioactive DNA was synthesized by supplying detergent-disrupted
virus preparations with magnesium and the deoxyrjboside triphosphates,
with one of them being labeled with tritium. When the synthesis
is completed, the protein and the RNA present are eliminated, and
the residual radioactive DNA is purified to completion. Each [³H]DNA
preparation is then rigorously examined for specific hybridizability
to its appropriate template and for its inability to complex with
irrelevant RNAs. After satisfying the specificity criteria, the
purified viral-specific tritiated DNA is mixed with cytoplasmic
RNA prepared from a variety of tumors and annealed under the conditions
described in Figure 1. The hybridizations are always carried out
with a vast excess of tumor RNA. Since the viral-specific tritiated
DNA is small compared with the RNA, any complexes formed between
them will behave physically more like RNA than DNA. Such complexes
are readily detected by isopynic separation in equilibrium density
gradients of cesium sulfate. At the end of the centrifugation, the
distribution of the tritiated DNA is examined across the gradient.
Any uncomplexed DNA will remain at a density corresponding to about
1.45. The DNA molecules that have annealed either partially or completely
to RNA will band at or near the density of RNA (epsilon = 1.65).
The movement of the tritiated DNA from the DNA density region to
the RNA density region is then the signal that the probe used has
found complementary sequences in the tumor RNA with which it is
being challenged.
MOLECULAR HYBRIDIZATION OF TUMOR p-RNA AND VIRAL SPECIFIC ³H-DNA
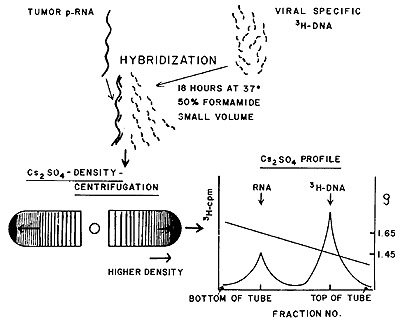
Fig. 1: Molecular hybridization and detection with viral-specific
[³H]DNA and tumor RNA (see text for further details).
The human neoplasias examined included adenocarcinoma of the breast
(2-4), the leukemias (5), the sarcomas (6), and the lymphomas (7).
The leukemias encompassed both acute and chronic varieties of the
lymphatic and myelogenous types. The human sarcomas studied included
fibro, osteogenic, and liposarcomas. The lymphoma series contained
Hodgkin's disease, Burkitt's tumors, lymphosarcomas, and reticulum
cell sarcomas. Control adult and fetal tissues were always examined
in parallel, and these were invariably negative. In the case of
breast tissue, the two benign diseases, fibroadenoma and fibrocystic
disease, were also included and were found to be negative.
Table II summarizes in diagramatic form the outcome of the survey
of human neoplasias with the animal virus probes. The pluses signify
that the corresponding tritiated DNA complexed with the indicated
tumor RNAs and the minuses, that no such complexes were detected.
The positives in these earlier studies ranged from 67 0/0 for breast
cancer to 92 0/0 for the leukemias. What is most noteworthy of the
pattern exhibited in Table II is its concordance with predictions
deducible from the murine system. Thus, human breast cancer contains
RNA homologous only to that of the murine mammary tumor virus. The
human leukemias, sarcomas, and lymphomas all contain RNA sharing
sufficient homology to that of the Rauscher murine leukemia virus
(RL V) to make a stable duplex. These mesenchymal neoplasias contain
no RNA homologous to the MMTV RNA. Finally none of the human tumors
contains RNA detectably related to that of the avian myeloblastosis
virus. The homology of leukemic RNA to that of RL V and the homology
of RNA from human breast cancer to that of MMTV have been confirmed
(8,9).
In summary, the specificity pattern of the unique RNA found in the
human neoplasias is in complete agreement with what has been described
for the corresponding virus-induced malignancies in the mouse.
Table II: Homologies among Human Neoplastic RNAs and Animal
Tumor Viral RNAs

The results of molecular hybridization between [³H]DNA complementary
to the various viral RNAs and pRNA preparations from the indicated
neoplastic tissues. The plus sign indicates that hybridizations
were positive and the negative sign, that none could be detected
(5).
IV. TheSimultaneous Detection Test
The existence of RNA in human tumors having sequence homology to
virus particles causing homologous diseases in mice does not of
course establish a viral etiology for these diseases in man. The
next step requires the performance of experiments designed to answer
the second and third questions raised in the introductory paragraphs,
i. .e., those relating to the size of the RNA being detected and
whether it is associated with the reverse transcriptase in a particle
possessing other features of complete or incomplete oncornaviruses.
What we sought was a method of detecting the presence of particles
similar to the RNA tumor viruses that would be simple, sensitive,
and sufficiently discriminating so that a positive outcome could
be taken as an acceptable signal of the presence of a viral-like
agent. To achieve this goal, we devised a test that depended on
the simultaneous detection of two diagnostic features of the animal
RN A tumor viruses.
The oncornaviruses exhibit two identifying characteristics. They
contain a large (1 x 107 daltons in molecular weight and composed
of subunits each of which is 3 x 106 daltons) single-stranded RNA
molecule having a sedimentation coefficient of 705, or 355 if the
705 molecule has broken down into its subunits. They also have reverse
transcriptase (10, 11), an enzyme that can use the viral RNA as
a template to make a complementary DNA copy.
The possibility of a concomitant test for both the enzyme and its
template was suggested by our prior experience with RNA transcriptase
in which we found (12) that the growing RNA chain could be detected
as a complex with its DNA template on removal of the protein from
the reaction mixture. Similar observations were made in examinations
of the early reaction intermediate (13, 14) of the reverse transcriptase
reaction.
I t was on this basis that 5chlom and 5piegelman ( 15) developed
the simultaneous detection test that was used to demonstrate ( 16)
the presence in human milk of particles containing 705 RNA and the
reverse transcriptase. The test was modified (17) to be applicable
to tumor tissue using the mouse mammary tumor as the experimental
model.
Figure 2 diagrams the procedure used. Tumor cells are first broken
by the use of the Dounce homogenizer and nuclei, mitochondria, and
large cell membrane fragments removed by low speed centrifugation.
The supernatant is subjected to trypsin digestion to inactivate
any nucleolytic enzymes and the trypsin is neutralized by trypsin
inhibitor. The supernatant is then centrifuged at 150,000 X g to
yield a cytoplasmic pellet containing virus particles, if present.
The resulting pellet is then banded isopycnicly in a sucrose density
gradient and the fraction between 1.16 and 1.19 g/ml is collected
by centrifugation. The recovered pellet is then treated with a nonionic
detergent (NP40) to disrupt possible viral particles, and the disrupted
preparation is used in a brief endogenous reverse transcriptase
reaction. The product of the reaction, with its RNA template, is
freed of protein and analyzed in a glycerol velocity gradient to
determine the sedimentation coefficient of the tritiated DNA. In
addition, the product is subjected to equilibrium centrifugation
in a Cs2SO4 gradient to determine its density.
The presence of particles encapsulating 705 RNA and a reverse transcriptase
will be indicated by the appearance of a peak of newly synthesized
DNA traveling at a speed corresponding to either a 705 RNA or a
355 RNA molecule. That the apparently large size of the [³H]DNA
is due to its being complexed to an RNA molecule can be readily
verified by subjecting the purified nucleic acid to ribonuclease
prior to velocity examination. The disappearance of the 705 and
355 [³H]DNA peaks following RNase treatment proves that the
[³H]DNA was complexed to large RNA molecules. Similarly, if
the reaction is positive, newly synthesized DNA should appear in
the RNA and/or hybrid regions of the Cs2S04 gradient, and these
peaks should again be eliminated by prior treatment with ribonuclease.
The simultaneous detection test was first applied to human breast
cancer (18) in a series including 38 adenocarcinomas and ten non-malignant
controls. It was found that 79 0/0 of the malignant samples were
positive for the simultaneous detection reaction and all of the
control samples from normal and benign tissue were negative. It
was further shown that the particles possessing the reverse transcriptase
activity and its 70S RNA template localize at a density between
1.16 and 1.19 g/ ml, the densi ty characteristic of the oncogenic
viruses.
The data obtained therefore indicate that one can, with a high probability,
find in human breast cancers particulate elements of the right density
that encapsulate RNA-instructed DNA polymerase and a 70S RNA.
SIMULTANEOUS DETECTION OF PARTICULATE RNA AND REVERSE TRANSCRIPTASE
IN CELLS
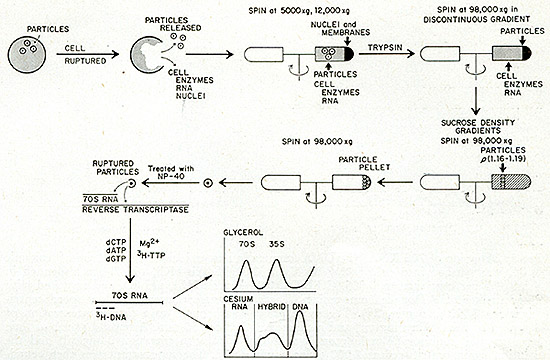
Fig. 2: Simultaneous detection test for 70S RNA and reverse
transcriptase in neoplastic tissue (see text for further details).
V. Application of the Simultaneous Detection Test to Mesenchymal
Tumors
In our initial study of the leukemias (19), peripheral leukocytes
were prepared from the buffy coats of both leukemic and nonleukemic
control patients. Cells were disrupted and fractionated as described
in Figure 2. Representative experiments examining the effects of
ribonuclease treatment of the product and omission of one of the
deoxytriphosphates during the reaction are shown in Figure 3. We
see the telltale 70S peaks of DNA synthesized by the pellet fractions
from the leukocytes of patients with acute lymphoblastic and acute
myelogenous leukemias. The elimination of the complex by prior treatment
with ribonuclease (Figure 3A) shows that the tritiated DNA is indeed
complexed to a 70S RNA molecule. Further, the omission of dATP (Figure
3B) leads to a failure to form the 70S complex, a result expected
if the reaction is in fact leading to the synthesis of a proper
heteropolymer. In similar experiments, it was shown that omission
of either dCTP or dGTP also resulted in the absence of the 70S RNA-[³H]DNA
complex, all of which argues against nontemplated end addition reactions.
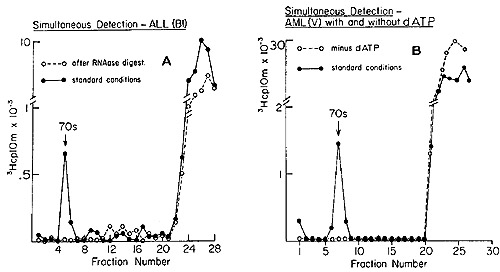
Fig. 3: Detection of 70S RNA [³H]DNA complex in human
leukemic cells.l gm of leukemic WBC was washed in 5 ml of 0.01 M NaCl,
0.01 M Tris-HC1, pH 7.4, resuspended in 4 ml of 5 per cent sucrose,
0.005 M EDTA, 0.01 M Tris-HC1, pH 8.3, and ruptured with three strokes
of a Dounce homogenizer. The nuclei were removed by low speed centrifugation
(2,000 g, 5 min, 2°). The supernatant was brought to a final concentration
of 1 mglml trypsin (Worthington) and incubated at 37° for 30 min.
A tenfold excess of lima bean trypsin inhibitor (Worthington) was
added (final concentration 3 mglml) and the solution again centrifuged
at 2,000 g for 5 min at 2°. The supernatant was then centrifuged
at 45,000 rpm for 60 min at 2°. The resulting cytoplasmic pellet
was resuspended in 0.5 ml of 0.01 M Tris-HC1, pH 8.3, brought to 0.1
per cent Nonidet P-40 (Shell Chemical Co.) and incubated at 0°
für 15 min. DNA was synthesized in a typical reverse transcriptase
reaction mixture (final vol 1 ml) containing: 50 µmol of Tris-HC1,
pH 8.3, 20 µmol NaCl, 6 µmol MgC12, lOO µmol each
of dATP, dGTP, dCTP, and 50 µmol[³H]dTTP (Schwarz Biochemical,
800 cpm per pmol). 50 µglml actinomycin D were added to inhibit
DNA-instructed DNA synthesis. After incubation at 37° for 15 min,
the reaction was adjusted to 0.2 M NaCl and 1 per cent SDS, and deproteinized
by phenol-cresol extraction. The aqueous phase was layered on a 10
to 30 per cent gradient of glycerol in TNE buffer (0.01 M Tris-HC1,
pH 8.3, 0.1 M NaCl, 0.003 M EDTA) and centrifuged in a SW-41 rotor
Spinco at 40,000 rpm for 180 min at 2°. Fractions were collected
from below and assayed for TCA-precipitable radioactivity. In this,
as in all sedimentation analysis, 70S RNA of the avian myeloblastosis
virus was used as a marker.
(A) One aliquot of product was run on the gradient as a control and
the other was pretreated with 20 µg of RNase 1 (Worthington)
for 15 min at 37° prior to sedimentation analysis. (B) Reactions
with and without dATP (19).
In some cases leukemic cells were obtained in amounts adequate to
permit a more complete characterization of the product. Hybridization
of the human product to the appropriate viral RNAs provides the
most revealing information since it tests sequence relatedness to
known oncogenic agents. We summarize in Table III the results of
examining the peripheral leukocytes of 23 patients, all in the active
phases of their disease, including both acute and chronic leukemias.
Of the 23 leukemic patients examined, 22 showed clear evidence that
their peripheral leukocytes contained particles mediating a reaction
leading to the appearance of endogenously synthesized DNA in the
70S region of a glycerol gradient. Nine of these were tested for
ribonuclease sensitivity and in all cases the complexes were destroyed.
In nine others, the DNA was recovered from the complex and annealed
to RLV RNA and
Table III: Simultaneous Detection of 705 RNA and Reverse Transcriptase
in Leukemic Cells (19)
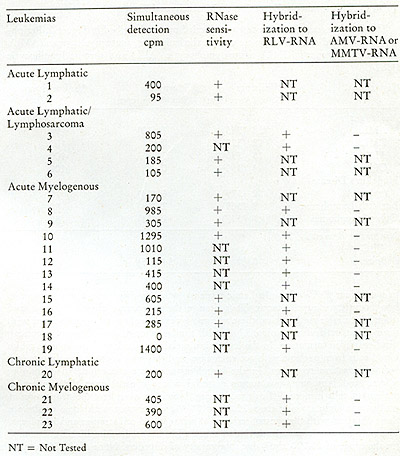
to either MMTV RNA or AMV RNA. In all nine, hybridizations occurred
with RL V RNA and not to either of the unrelated MMTV RNA or AMV RNA.
In four patients, enough DNA complex was formed to permit a complete
characterization of the product. In all four, the DNA complexes were
destroyed by ribonuclease and the purified DNA hybridized uniquely
to RL V RNA.
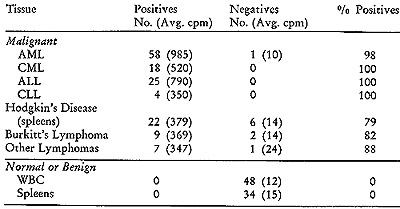
In addition to this initial group, we subsequently examined 85 leukemic
patients and 38 patients with lymphomas (20, 21), including Hodgkin's
disease, African Burkitt's lymphoma, lymphosarcoma, and reticulum
cell sarcoma. The results of the simultaneous detection tests on these
and corresponding control tissues are summarized in Table IV. I t
is noteworthy that positive outcomes were observed in more than 99
% of the leukemic patients, whether they were acute or chronic, lymphocytic,
or myelogenous. Thus despite their disparate clinical pictures and
differing cellular pathologies, these various types of leukemias are
associated with virus-like particles containing RNA with similar,
though probably not identical, viral-related information. In the leukemias,
we always dealt with peripheral white blood cells from patients with
active disease, and this may account for almost total lack of negative
responses. In the lymphomas, we were confined to examining spleens
and lymphomatous tumor material where control over the content of
malignant cells is more difficult to exercise. However, even here
the proportion of positives is high, ranging from 79 % to 88 %. In
contrast with these results are those obtained with the control series
of 48 white blood cell samples and 34 spleens. The non-neoplastic
samples included some with elevated white blood cell counts (in the
range of 25,000/mm³) due to a variety of disorders. None of the
82 samples from cancer-free patients exhibited any evidence of positive
reactions. The difference in average cpm of positives and negatives
is such that a diagnostic decision is unambiguous.
Table IV: Simultaneous Detection on Mesenchymal Tissues
The simultaneous detection test was carried out as described
in Figure 2. Peripheral white blood cells (WBC) were used in the
leukemias, acute myelogenous (AML), chronic myelogenous (CML), acute
lymphocytic (ALL) and chronic lymphocytic (CLL).
VI. Implications of Simultaneous Detection Tests on Human Breast
Cancer and the Mesenchymal Tumors
The experiments we have just summarized on human breast cancer and
the leukemias were designed to probe further the etiological significance
of our exploratory investigations (2, 5), which identified in these
neoplasias RNA homologous to those of the corresponding murine oncornaviruses.
The data obtained with the simultaneous detection test established
that at least a portion of the tumor-specific virus-related RNA we
were detecting was a 70S RNA template physically associaed wi th a
reverse transcri ptase in a particle possessing a densi ty between
1.16 and 1.19 g/ml, three of the diagnostic features of the animal
RNA tumor viruses. Further, the DNA synthesized in the particles from
both classes of neoplasias hybridized uniquely to the RNA of the corresponding
oncornavirus. Note that this last result is complementary to and completes
the logic of our experimental approach. We started out by using animal
tumor viruses to generate [³H]DNA probes that were used to find
related RNA in human neoplastic tissue. We concluded by using analogous
human particles to generate [³H]DNA probes, which were then used
to determine sequence relatedness to the RNA of the relevant oncornaviruses.
None of the human probes hybridized to the avian viral RNA. The probe
generated by the particles from human breast cancer was homologous
only to the RNA of mouse mammary tumor virus, whereas the human leukemic
probe was related in sequence only to RL V RNA, the murine leukemic
agent. The biologically logical consistency of these results adds
further weight to their probable relevance to the human disease.
VII. On the Problem of Germ-line Transmission of Viral Information
We now come to grips with the fifth question raised in the introductory
paragraphs, the virogene-oncogene concept (22), which derives from
animal experiments and argues that all animals prone to cancer contain
in their germ line at least one complete copy of the information
necessary and sufficient to convert a cell from normal to malignant
and produce the corresponding tumor virus. This hypothesis presumes
that the malignant segment normally remains silent and that its
activation by intrinsic or extrinsic factors leads to the appearance
of virus and the onset of cancer .
There are various ways of testing the validity of the virogene-oncogene
hypothesis, but the pathways differ in the technical complexities
entailed. One approach commonly used attempts to answer the question:
Does every normal cell contain at least one complete copy of the
required viral-related malignant information? The methodologies
used included the techniques of genetics, chemical viral induction,
and molecular hybridizations. However, for a variety of reasons,
none of these gave, or could give, globally conclusive answers.
Genetic experiments do not readily distinguish between susceptibility
genes and actual viral information. Further, even if genetic data
succeeded in identifying some structural viral genes, it would still
be necessary to establish that all the viral genes are represented
in the genome. Attempts to settle the question by demonstrating
that every cell of an animal can be chemically induced to produce
viruses have thus far, for obvious reasons, not been tried. The
best that has been achieved along these lines is to show that cloned
cells do respond positively. However, the proportion of clonable
cells is small and clonability may well be a signal for prior infection
with a tumor virus.
Finally, the quantitative limitations of molecular hybridization
make it almost impossible to provide definitive proof that each
cell contains one complete viral copy in its DNA. Although it is
not very difficult to show that 90 0/0 of the information is present,
it is the last 10 0/0 that constitutes the insurmountable barrier
and 10% of 3 x 10 high 6 daltons amounts to a far from trivial 3
x 10 high 5 daltons, the equivalent of about one gene.
A useful way to obviate these technical difficulties is to invert
the problem. Instead of asking whether one complete copy exists
in normal cells, the question can be phrased in the following terms:
Does the DNA of a malignant cell contain viral-related sequences
that are not found in the DNA of its normal counterpart? Phrasing
the issue in this manner leads to the design of experiments that
avoid the uncertainties generated by the demonstrated fact that
many indigenous RNA tumor viruses share, completely or partially,
some sequences with the normal DNA of their natural hosts (23 ).
The crucial point is of course whether all of the viral sequences
are to be found in normal DNA. The approach we adopted requires
removal of those viral sequences that are contained in non-neoplastic
DNA by exhaustive hybridization of the viral probe to normal DNA
in vast excess. Any unhybridized residue can then be used to determine
whether malignant DNA contains viral-related sequences not detectable
in normal tissue.
We first investigated this question in the case of the human leukemias
(24) and the strategy, as diagrammed in Fig. 4 (a and b), may be
outlined in the following steps:
a) Isolate from leukemic cells the fraction enriched for the particles
encapsulating the 70S RNA and RNA-directed DNA polymerase;
b) Use this fraction to generate [³H]DNA endogenously synthesized
in the presence of high concentrations of actinomycin D to inhibit
host and viral DNAdirected DNA synthesis;
c) Purify the [³H]DNA by hydroxyapatite and Sephadex chromatography
with care being exercised to remove by self-annealing and column
chromatography all self-complementary material in the tritiated
probe;
d) Use the resultant [³H]DNA to detect complementary sequences
in normal and leukemic leukocyte DNA;
e) If viral-related sequences are detected in both, remove those
found in normal leukocytes by exhaustive hybridization to normal
DNA; and
f) Test the residue for specific hybridizability to leukemic DNA.
In carrying out the recycling and test hybridizations, it is imperative
that conditions be chosen to account for the possibility that the
leukemia-specific sequences are present in only one copy per genome,
a possibility which is in fact realized (24). To this purpose, the
concentration in moles per liter (Co) of DNA and the time (t in
seconds) of annealing is adjusted to Cot values of 10,000, which
are adequate to locate unique sequences.
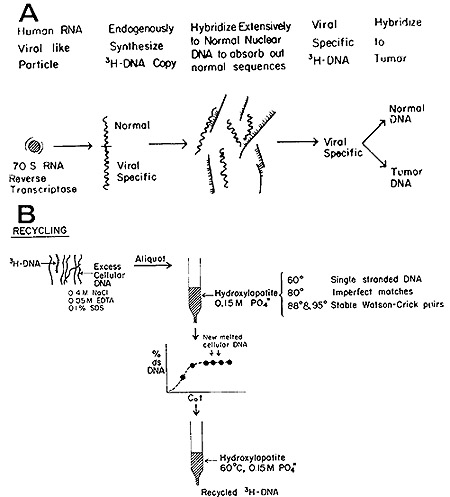
Fig. 4: (A) Generation of [³H]DNA by human leukemic
particles and hybridization of sequences shared with normal DNA. (B)
Separation of leukemia-specific sequences by hydroxyapatite chromatography.
See text for further details.
A typical outcome of hybridizing such recycled tritiated DNA to
normal and leukemic DNA is shown in Fig. 5. It is evident that no
complexes stable at temperatures above 88° are formed with normal
DNA. On the other hand, 57 0/0 of the recycled [³H]DNA probe
forms well-paired duplexes with leukemic DNA. A series of such experiments
was performed with particle-generated [³H]DNA and nuclear DNA
obtained from 8 untreated patients with either acute or chronic
myelogenous leukemia. In every case (Table V), the [³H]DNA,
after being sub-
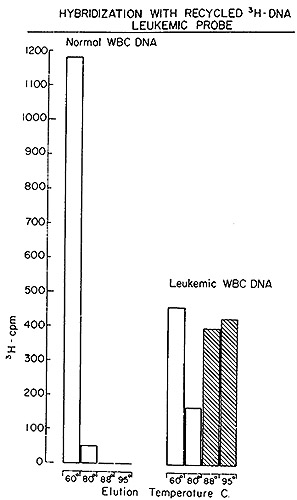
Fig. 5: Hydroxyapatite elution profile of a hybridization
reaction of recycled leukemie [³H]DNA to nuclear DNA from normal
leukocytes and from leukemic leukocytes of the patient from which
the [³H]DNA was derived.
jected to exhaustive annealing to normal DNA, yielded a residue
that forms stable duplexes only with leukemic DNA, in agreement
with the experiment of Fig. 5.
In estimating the implication of these results, it must be recalled
that the leukemia-specific sequences found (24) in leukemic cells
are present as non reiterated copies per genome. This was established
by the Cot values ( concentration of nucleotides X time) required
to detect them. The sensitivity used to examine normal cells for
the leukemia-specific sequences was such that l/50th of an equivalent
of that found in leukemic cells would have been readily detected.
Consequently, one may conclude that the vast majority of normal
cells do not contain this particular stretch of malignant-associated
information and it cannot therefore be represented in the germ line
of nonleukemic individuals.
Table V:
Exhaustive Hybridization of [³H]DNA Probe Synthesized by Leukemic
Particles with Normal-leukocyte Nuclear DNA, Followed by Hybridization
of the Nonhybridizing Recycled Leukemic [³H]DNA Probe to Normal
DNA and to Leukocyte Nuclear DNA from the same Leukemic Patient (24).
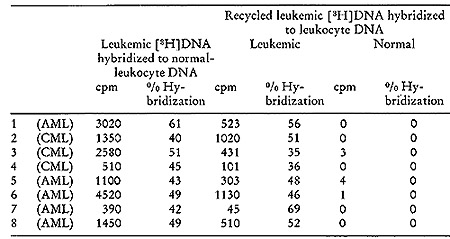
Background was 30 cpm and all counts recorded represent cpm above
background.CML = Chronic meyelogenous leukemia. AML = Acute myelogenous
leukemia.
VIII. Unique Sequences in Hodgkin's and Burkitt's Lymphomas
and their Relatedness
We have already noted that, like the leukemias, Hodgkin's and Burkitt's
lymphomas have particles containing reverse transcriptase and a
70S RNA template related in sequence to that of RL v. It was of
obvious interest to determine whether the lymphomas also parallel
the leukemias in possessing a unique sequence not detectable in
normal tissue. If they do, one can in addition ascertain whether
the sequences found in Hodgkin's and Burkitt's lymphomas are related
to each other. The outcome has evident significance for the possible
relevance of the sequence to malignancy.
[³H]DNA probes were synthesized with particles isolated from
four Burkitt's tumors and three Hodgkin's disease specimens. Sequences
shared with normal DNA (between 35010 and 400/0) were then removed
as described for the leukemias (24) to yield the recycled [³H]DNA
probes (25).
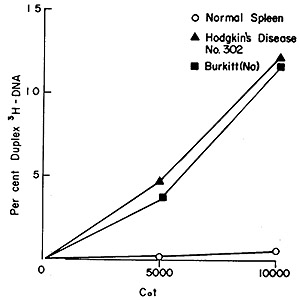
Fig. 6: Hybridization of recycled Hodgkin's disease #302
(³H]DNA to nuclear DNA isolated from normal spleen (0-0), Hodgkin's
disease #302 (black triangle -black triangle), and Burkitt's lymphoma
(NA) (black square -black square). Equal aliquots were removed from
the hybridization vessel at each Cot value and hybrid formation was
analyzed by hydroxyapatite chromatography. The input counts for each
point were 1,500-2,000 cpm and only those duplexes eluting at 88 °
and above are counted as stably hybridized.
Figure 6 shows the outcome of challenging recycled Hodgkin's disease
[³H]DNA with nuclear DNAs from normal spleen, Hodgkin's disease
spleen, and from Burkitt's lymphoma. Few, if any, stable duplexes
are formed with normal DNA. Note, however, that although the probe
was made with Hodgkin's disease particles, the [³H]DNA hybridized
to Burkitt's lymphoma nuclear DNA virtually as well as it complexed
to DNA from Hodgkin's spleen. The converse is also true, as may
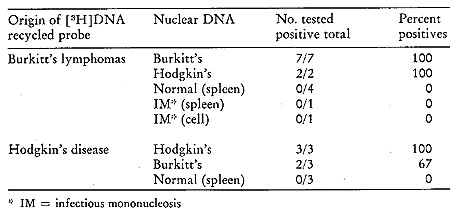
be seen from Table VI, which summarizes the results of our findings
in the recycled [³H]DNA challenged with nuclear DNA from normal
and malignant tissues (25 ). Again, normal DNA is unable to form
significant amounts of stable complexes (elution at 88° and
above) with the [³H]DNA probes. In all instances, the lymphoma
[³H]DNAs hybridized in stable complexes to the nuclear DNA
of the original types from which the particles were obtained and
used to generate the labeled DNA. Further, with only one exception,
all of the Burkitt's and Hodgkin's disease [³H]DNAs cross hybridize
with each other's DNA.
In summary, several features emerged from this study of the lymphomas.
The particle-related sequences found in Burkitt's and Hodgkin's
lymphomas possess sequences in common, an observation in accord
with our earlier findings (7, 20, 21, 26 ), that Hodgkin's and Burkitt's
particles both share sequences with the Rauscher murine leukemia
agent. Further, in view of the previous association of the Epstein-Barr
virus with Burkitt's lymphoma (27, 28) and the non-neoplastic infectious
mononucleosis (29, 30), it is revealing to note from Table VI that
the leukocyte DNA of patients with infectious mononucleosis was
devoid of the Burkitt's sequences detected by the recycled [³H]DNA
lymphoma probe, indicating that these latter sequences are specific
for neoplastic tissues. The fact that the particle-related sequences
in Hodgkin's and Burkitt's tumors are related to each other adds
further weight to this conclusion. Finally, the observation that
cells carrying multiple copies of the DNA of the Epstein-Barr virus
do not complex with recycled [³H]DNA probes from either Hodgkin's
or Burkitt's particles proves that these particle sequences have
no detectable relation to the DNA of the Epstein-Barr virus.
Table VI:
Hybridization of Recycled [³H]DNA Probes Synthesized with Human
Lymphoma Particles
with Nuclear DNA from Normal and Tumor Tissues
IX. Evidence from Studies of Identical Twins
Although the comparison of leukemic patients with normal suggests
that healthy individuals do not contain the leukemia-specific sequences,
the data do not rule out the possibility that those who do come
down with the disease do so because they in fact inherit the required
information in their germ line. One way to resolve this issue is
to study the situation in identical twins. Since identical twins
are monozygous, i. e., derive their genomes from the same fertilized
egg, any chromosomally transmitted information must be present in
both. It had already been shown by Goh and his colleagues (31, 32)
in the case of chronic myelogenous leukemia that only the leukemic
member of each of two identical twin pairs contained the marker
Philadelphia chromosome. It was of obvious interest to examine this
situation for the leukemia-specific sequences. If the leukemic member
of the pair contains the particle-related DNA sequences, and does
so because he inherited them through his germ line, then these same
sequences must be found in
the leukocyte DNA of his healthy sibling. To perform the experiment,
it was necessary to locate identical twins with completely convincing
evidence for monozygosity and where only one of them was leukemic.
Further, the twins had to be of adult age since at least a unit of
whole blood is required to provide enough leukocyte DNA to carry out
the required hybridization.
Two sets of identical twins satisfying all these requirements were
found and an experiment similar to the one outlined above was performed
with each pair (33). In each instance, particles containing the reverse
transcriptase and 70S RNA were again isolated from the leukocytes
of the leukemic members and used to generate the [³H]DNA endogenously.
The [³H]DNA was purified and sequences shared with normal DNA
removed by exhaustive hybridization in the presence of avast excess
of normal DNA from random healthy blood donors. This was then followed
by hydroxyapatite chromatography to separate paired from unpaired
[³H]DNA. It is important to emphasize that in the recycling step,
the normal DNA used came from the leukocytes of healthy, random blood
donors and not from the normal twin. To have used the latter would
have obviously confused the issue. The residue of the tritiated DNA
that did not pair with the normal DNA was then used to test for the
presence of a sequence in the leukocyte DNA of the patient and that
of his healthy sibling.
The results obtained with the two sets of twins are described in Fig.
7, and it is evident that the same situation holds between the members
of the twin pairs as was observed in the comparison of unrelated leukemic
patients and random normals (Fig. 5 and Table V). The leukemic twin
contains particle-related sequences that cannot be detected in the
leukocytes of his healthy sibling.
The fact that we could establish a sequence difference between identical
twins implies that the additional information found in the DNA of
the leukemic members was inserted after zygote formation. This finding
argues against the applicability of the virogene hypothesis to this
disease since it would demand that the leukemia-specific sequences
found in the DNA of the individual with the disease must surely also
exist in the genome of his identical twin. These results are also
inconsistent with the possibility that individuals who succumb to
leukemia do so because they inherit the complete viral genome.
X. Implications of the Unique DNA Sequences in Leukemias and
Lymphomas
The data we have summarized on the existence of virallateral-related
sequences unique to the DNA of human malignant cells imply that
they are inserted in somatic DNA, a process known to occur with
RNA tumor viruses in animal cells in tissue cultures (34) and in
whole animals (35). The fact that these viral sequences can be incorporated
into somatic DNA suggests that this could also occur in early embryogenesis
and thus involve a cell destined to differentiate into the germ
line. An event of this nature would be selected for in any attempts
at developing inbred strains characterized by high frequency of
cancer .
Indeed this seems to have occurred in the course of producing the
AKR mouse, a strain in which spontaneous leukemia occurs with virual
certainty. It has been shown (36) that the DNA of the AKR mouse
contains murine leukemia virus sequences that are not present in
the DNA of the NIH Swiss mouse. These sequences were localized by
genetic and molecular hybridization, and it was found that they
are either identical to or closely linked to the Akv-1locus.
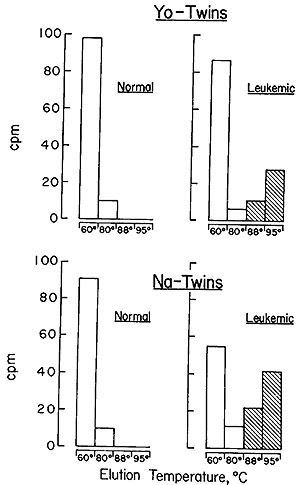
Fig. 7: Hydroxyapatite elution profile of a hybridization
reaction of the recycled leukemic twin [³H]DNA probe to nuclear
DNA from normal leukocytes, normal twin leukocytes, and leukocytes
from the same leukemic twin. The annealing reaction mixtures contained
20 A260 units of cellular DNA, 0.004 pmol of [³H] DNA, and
15 µmol NaH2PO4 (pH 7.2) in a final vol of 0.01 ml. The reaction
was brought to 98 ° for 60 sec and 0.04 mmol of NaCI was added.
The reaction mixture was then incubated at 60° X 50 hr. The
reaction was stopped by the addition of 1 ml of 0.05 M NaHP04 (pH
6.8). The sample was then passed over a column of hydroxyapatite
of 20-ml-bed v01 at 60°. The column was washed with 40 ml of
0.15 M NaHP04 (pH 6.8) at 60°,80°,88°, and 95°.
Fractions of 4 ml were collected, the A260 of each fraction was
read, and the DNA was precipitated with 2 µg/ml of carrier
yeast RNA and 10 per cent trichloroacetic acid. The precipitate
was collected on Millipore filters, which were dried and counted.
In all cases, greater than 80 per cent of the nuclear DNA reannealed.
A background count of 8 cpm was subracted in all instances (33).
The case of the AKR mouse was very likely an inadvertent result
of its selection. An even more remarkable instance is the deliberate
insertion of viral sequences into the germ line (37). This was accomplished
by infection of preimplantation mouse embryos at the 4-8 cell stage
with the murine leukemia virus (MuL V) followed by reimplan.tation
in the uteri of surrogate mothers. Of 15 such animals born, one
developed lymphatic leukemia at 8 weeks of age. Molecular hybridizations
revealed leukemia-specific viral sequences in the DNA of all eight
different organs examined, whether they were of mesenchymal origin
or not. In agreement with our earlier findings (35), these sequences
were not found in normal mesenchymal tissue nor were they detected
in the DNA of non-target tissues in animals made leukemic by injection
of virus after birth.
In summary, except for strains deliberately inbred for high spontaneous
occurrence of disease, the mesenchymal neoplasias of mice and men
would appear tO have a similar underlying mechanism. In both instances
new viral-related sequences are found in the DNA of the malignant
cells and these are not found in the DNA of uninvolved tissues.
They are therefore not germinal unless one wishes to invoke a rather
unlikely specific elimination in the course of the differentiation
of every cell but the malignant one. Despite its implausibility,
this possibility should be exploited by testing for the leukemic-specific
sequences in the germ line DNA (sperm) of leukemic individuals.
It must be emphasized that conclusions as to the validity of the
virogene-oncogene hypothesis are only relevant to the particular
instances examined and cannot be generalized to any other viral-related
cancers even in the same animal, let alone to other species. In
any event, it is evident that our findings with respect to the human
mesenchymal tumors suggest more optimistic pathways for the control
of these diseases than would be available if the total information
were already in the genome. The data imply that we may not be forced
to master the control of
our own genes in, order to cope with these neoplasias.
The fact that the human particles possess sequences homologous to
those found in viral agents known to cause the corresponding neoplasias
in mice encourages the hope that they are relevant to human disease.
However, despite the considerable progress that can be recorded,
no definitive proof exists at the present writing that the virus-like
particles found in the human neoplasias are either viruses or etiologic
agents of the cancers in which they are found. Proof will ultimately
come when it proves possible to produce the relevant malignancy
in a susceptible animal by injection of the particles purified from
human tumors. It should, however, be noted that we have known of
the mouse mammary tumor virus for more than 35 years and no one
has yet succeeded in producing mammary tumors with this agent in
any animal other than the mouse.
Under the circumstances, it would seem prudent not to wait for the
definitive experiment with the human particles, but rather to proceed
with attempts at further exploration of their significance and possible
clinical usefulness. We should like to list a few areas of possible
exploitation and then turn our attention to a brief description
of what has been accomplished along these lines.
1. The existence of the leukemia-specific sequences can provide
the clinician with a hitherto unsuspected parameter that could potentially
be a useful adjunct in monitoring therapy.
2. One could attempt to grow the human particles in tissue culture
in order to provide a more accessible source of these particles
for further biochemical characterization with the ultimate hope
of generating useful reagents for diagnostic, therapeutic or monitoring
purposes.
3. Another, less ambitious approach is to purify one of the protein
subcomponents from the human particles for further characterization.
This could then be used for the production of a monospecific antiserum
that might be clinically useful.
XI. Particulate Reverse Transcriptase in the Leukocytes of Leukemic
Patients in Remission
We have already noted (Table V) that positive simultaneous detection
tests, indicating the presence of particles containing reverse transcriptase
and the 70S RNAtemplate, were obtained in more than 99°% of
the leukemic patients examined. It was of obvious interest to see
whether these particles could be detected in the leukocytes of leukemic
patients who are in good clinical remission.
Peripheral blood leukocytes were obtained from patients at the Baltimore
Cancer Research Center and from the M. D. Anderson Hospital. The
leukocytes from some of the leukemic patients were obtained by leukophoresis
and immediately stored at -7° until used. The clinical statuses
of the patients at the time of leukophoresis are summarized in Table
VII. A complete remission was defined as the absence of symptoms
related to the disease, normal results on physical examination,
a hemoglobin of greater than 10 g/100 ml, leukocyte count greater
than 3000/mm³, platelet count greater than 100,000/mm³,
no blasts in the peripheral blood smear, and less than 5 010 blasts
in the bone marrow.
Table VIII summarizes the results of simultaneous detection assays
for high molecular weight RNA and reverse transcriptase in the leukocytes
from the patients examined. Outcomes are designated as positive
only when the peaks of tritiated DNA found in the 705 and 355 regions
were eliminated by prior treatment with ribonuclease, a feature
establishing that the [³H]DNA is complexed to a large RNA molecule.
If the peaks are not removed subsequent to RNase digestion, the
reaction is scored as a negative outcome. In the present study two
untreated leukemic patients were available for testing prior to
remission induction and both were positive at that time. Three of
the nine patients in complete remission demonstrated a 70S or 35S
peak of acid-precipitable radioactivity that was abolished by RNase
treatment. The "negatives" were subjected to a simultaneous
detection assay via a cesium sulphate gradient, a procedure that
obviates the problem generated by fragmentations of the RNA template
during manipulation.
It will be noted from Table IX that samples from the untreated patients
were all positive. The [³H]DNA-RNA hybrids were detected in
nine out of eleven patients in complete remission. In this group,
six of seven AML patients and two of three acute lymphocytic leukemia
(ALL) patients demonstrated a positive reaction. In five of the
patients, the simultaneous detection tests were negative by velocity
sedimentation analysis but were positive when analyzed in the cesium
Table VII:
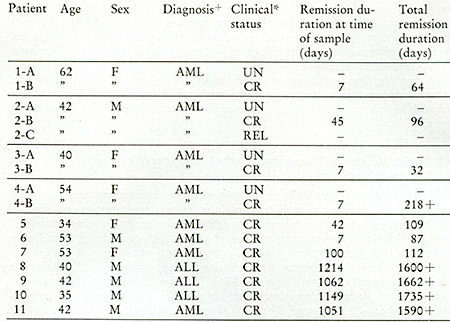
+AML = acutc myelogenous leukemia; ALL = acute lymphocytic leukemia
*UN = untreated; CR = complete remission; REL = re]apse
Clinical status of leukemic patients when leukophoresis was performed
for enzyme studies.
Table VIII:
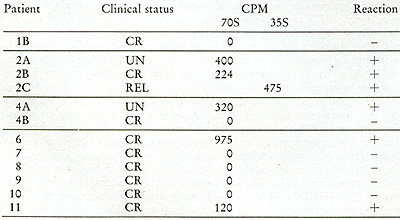
Test for 70S and 35S RNA-[³H]DNA in leukocytes from leukemic
patients. CPM in 70S or 35S represents acid-precipitable radioactivity
that was removed from the 70S and 35S by prior treatment with ribonuclease
A and T 1.
Table IX:
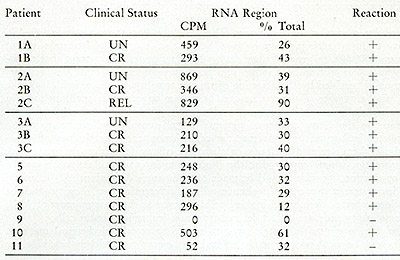
Cesium sulfate analysis of RNA[³H]DNA in leukocytes from leukemic
patients. The CPM in the RNA or RNA-DNA hybrid region and the percent
of the total CPM applied to the gradient are enumerated. A positive
reaction represents heat and ribonlicleasesensitive acid-precipitable
radioactivity in the RNA or hybrid region of the gradient as described
in the text. The [³H]DNA found in the hybrid density regions
is hybridized to smaller DNA-RNA complexes, which would result from
fragmentation of the larger 705 and 355 RNA molecules.
sulphate gradients. Hybrids of small molecular size would not be
identified as 70S with 35S complexes, but can be detected as complexes
in the hybrid region of the cesium sulphate gradient. Only one of
the patients (#9) was negative by both glycerol and cesium sulphate
gradient analyses. This patient did not appear to differ clinically
at the time of the examination from the other remission patients
exhibiting positive reactions. In the course of these studies we
also examined by cesium sulphate analysis two pooled, normal white
blood cell samples and five non-neoplastic spleens for the presence
of particles capable of yielding RNAtritiated DNA hybrids in an
endogenous reaction and all were negative, as had been true in our
previous studies (Table V).
It is obvious that finding the leukemia characteristic particles
in the white blood cells of patients in remission is disappointing
and does not accord with the generally accepted assumption that
there is a normal and a leukemic population of leukocytes in acute
leukemia (38). The goal of contemporary chemotherapy and immunotherapy
is to reduce the size of the leukemic component (to zero if possible)
to allow the bone marrow and the peripheral blood to repopulate
with non-neoplastic cells. A number of clinical observations suggest
that remission leukocytes are in fact normal cells. First, the prolongation
of life is directly proportional to the duration of the remission.
Secondly, a small but increasing number of patients with acute lymphoblastic
leukemia in long-term remission appear to go on to cure, indicating
a permanent extinction of the leukemic cell population (39).
The morphology and functional properties of remission leukocytes
have been studied by a number of techniques. These include karyotype
analysis (40-45), ability to form colonies in agar ( 46-49), and
the detection of leukemia-related antigens (50, 51). In general,
these studies have supported the concept that remission leukocytes
represent the return of a population of normal cells. Also, relapses
are usually heralded by the detection of the abnormality associated
with leukemic cells, and a number of these techniques have been
suggested as ancillary tools in following the response of patients
to chemotherapy. However, there have been instances in the above
reports of patients in well-consolidated remissions whose peripheral
leukocytes or bone marrow cells demonstrated persistence of aneuploidy,
leukemia-related antigens, or abnormal colony formations in agar;
these abnormalities appear unrelated to the effects of maintenance
of chemotherapy. In this connection, mention should be made of Killmann's
deductions (52) based on the demonstrated capacity of leukemic cells
to differentiate; on these grounds he questions whether the normal-looking
cells observed in the bone marrow of AML patients in remission are
in fact derived from non-leukemic ancestor cells.
The data presented indicate that with regard to certain biochemical
markers, which may be virus-related, remission leukocytes may more
closely resemble the leukemic cells than normal cells. Quite surprisingly,
two of the three ALL patients in long-term remission still had evidence
of particles in their peri pheralleukocytes. Further, the enzyme
was detected in seven of eight patients with AML in remission. We
are unable to determine if all or a fraction of the peripheral white
cells studied possessed the leukemic characteristics. Thus we cannot
directly answer the question whether one or two white cell populations
are present.
Mak and his colleagues (53, 54) have described particulate activity
in the supernatants of short-term cultures derived from bone marrow
of leukemic patients in remission. The activity of the cultures
from remission patients equaled, and in some instances exceeded,
that detected in the cultures derived from patients in relapse.
There are a number of plausible explanations for the persistence
of the particulate enzyme and its associated template in the remission
leukocytes. The normal cell found in remission could be infected
with a non-oncogenic C-type virus or conversely the remission leukocyte
could have acquired resistance to transformation whereas susceptibility
to infection was unaltered. Second, as a result of chemotherapy,
a portion of the leukemic clone could have evolved into a non-neoplastic
clone still capable of expressing some viral function. There are
a number of in vitro models for the latter phenomenon. Thus, it
has been shown that cells transformed with a murine sarcoma virus
can spontaneously, or after exposure to antimetabolites, revert
to a normal morphology. Certain clones of these morphological revertants
behave in a non-malignant manner, yet some viral functions are expressed
or can be induced (55).
A more direct method for examining such questions is to use the
molecular hybridization to answer the following questions: 1) Do
remission cells have leukemia-specific DNA nucleotide sequences?
2) If present, are some leukemia-specific DNA sequences not expressed
or are critical DNA sequences deleted? These areas are presently
under investigation.
XII. Attempts to Produce Human RNA Tumor Particles in Cell Cultures
All would agree that an important advance would result from the establishment
of particle-producing cells in short, or preferably long-term culture.
An alternative but equally useful outcome could be obtained by the
successful infection of established cell lines with the human virus-Iike
particles. Although not yet achieved, a number of recent reports suggest
that this desirable situation may eventually be obtained. Thus, McGrath
et al. ( 56) describe a human breast carcinoma cell line that may
ultimately be converted into a source of breast cancer particles.
Kotler et al. (57) have succeeded in using arginine starvation to
induce the release of virus-Iike particles from human leukemic cells.
We have already noted that shortterm cultures of leukemia bone marrow
aspirates in a conditioned medium has led to the production of particles
recoverable from the culture supernatants (53,54).
Probably the most interesting recent announcement along these lines
came from Gallo and his colleagues (58, 59, 60) who reported the isolation
of a C-type virus (HL23V) from cultured peripheral white blood cells
derived from a patient with acute myelogenous leukemia. The reverse
transcriptase of this putative human oncornavirus was found to be
antigenically related to the reverse transcriptase of the simian sarcoma
virus type-1 (SSV -1) and to the gibbon ape lymphoma virus (GAL V).
The spontaneously released viruses from the human leukemia cells were
successfully transmitted to A204, a human rhabdomyosarcoma cell line.
The infected A204 (HL23V) culture was an excellent producer, yielding
virus in sufficient quantities to permit biochemical and immunological
characterization.
The potential implications of these observations made it mandatory
to undertake the task of identifying the nature of the virus particles
released. We will here briefly summarize our efforts along these lines.
The A204 (HL23V) culture produced high titers of particles that were
found by (³H]-uridine labeling to possess the characteristic
buoyant density (1.16 g/ml) of oncornavirus. Simultaneous detection
assays (15) of the culture supernatants demonstrated that the particles
encapsulated 70S RNA and reverse transcriptase.
The reverse transcriptase from A204 (HL23V) culture supernatants was
examined for relatedness to the SSV enzyme. Figure 8 shows that the
antiserum prepared against the SSV reverse transcriptase was capable
of inactivating the reverse transcriptase activity of HL23V particles
only to about 60 %. The partial inhibition of enzyme activities suggested
the possible presence of a second virus containing an antigenic ally
unrelated enzyme.
To identify the unknown component in the HL23V particles, a search
was instituted amongst known oncornaviruses using immunologic and
molecular hybridization techniques. Probable candidates were quickly
narrowed down to the RD114/CCC baboon endogenous virus group. Figures
9A and 9B show hydroxyapatite temperature elution profiles of viral
(SSV and BV-M7) cDNA annealed to the total RNA from HL23V particles.
The extent of the hybridization and the thermal stability indicate
that the HL23V particles contain the complete information of both
the simian sarcoma virus (SSV) and the baboon endogenous virus (BV-M7).
To determine whether all of the genetic information of HL23V can be
accounted for by these two viruses, the reciprocal hybridization was
performed.
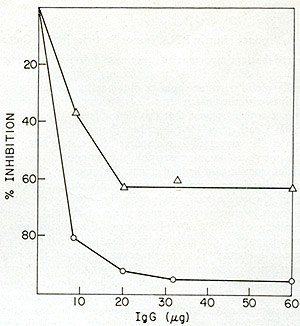
Fig. 8: Effects of anti-SSV reverse transcriptase IgG on
HL23 reverse transcriptase activity. Increasing amounts of IgG purified
from normal goat serum and from goat antiserum directed against SSV
reverse transcriptase were mixed with NP-40-disrupted HL23, incubated
for 15 min at 37° and then assayed for reverse transcriptase.
SSV was similarly treated and assayed. Both HL23 and SSV input were
standardized to incorporate 10 pmoles of [³H] -TP in a synthetic
template assay. The reverse transcriptase reactions (100 µl)
contained the following in µmoles: Tris-HCl (pH 8.0), 5; MnC12,
0.02; KCI, 4; dithiothreitol, 0.04; 0.02 each of dGTP and [³H]
-dGTP (500 cpm/pmole) and oligo dG12: poly Cm at 4 µg/ml. After
incubation at 37° for 30 min, the reactions were terminated and
assayed for acid-precipitable radioactivity. Using incorporations
at identical inputs of normal IgG as control, the percent inhibition
of the SSV and HL23 polymerase activity by the increasing levels of
immune IgG were computed. 0 = V; white triangle HL23 virus. the increasing
levels of immune IgG were computed. 0 = SSV; white triangle = HL23
virus.
In these experiments, cDNA probe synthesized endogenously with
HL23V particles, was annealed to RNAs from SSV-1 or BV-M7 or both.
Figure 10 shows that the HL23V-cDNA hybridized 37 % and 44 % to
the RNAs of BV-M7 and SSV-1, respectively. These individual hybridizations
were additive as demonstrated by the complete complexing of HL23V-cDNA
to a mixture of BV-M7 and SSV-1 RNAs. These data indicate that the
genetic information of HL23V virions is completely accounted for,
within the limits of the sensitivity of the molecular hybridization
technique used, by the complete genomes of both SSV-1 and BV-M7.
To supplement and confirm these findings by an independent method,
competition molecular hybridizations were performed using cDNA synthesized
from SSV. Viral RNA from SSV was labeled with 125I. SSV-cDNA can
protect this 125I
SSV-RNA more than 79 % from ribonuclease digestion at molar ratios
of 5:1 (cDNA:RNA). Table X shows that when unlabeled viral RNAs
were added in vast excess to compete this homologous reaction, both
SSV and HL23V RNA
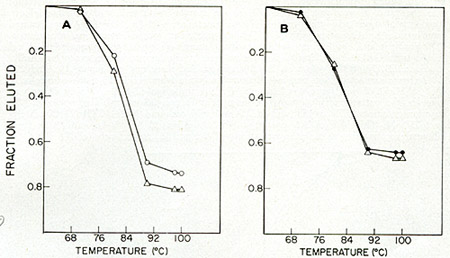
Fig. 9: Hybridization of [³H]-DNA transcripts
of (A) 55V-1 and (B) BV (M7 isolate) to HL 23 viral RNA. 55V-1 and
BV-M7 were obtained from Pfizer, Inc. (Maywood, N. J.) and were derived
from sucrose density gradient-banded tissue-culture supernatants of
a chronically infected human Iymphoblastoid cell line (NC-37) and
baboon kidney-canine thymus coculture (BKCT), respectively. HL23 virus
was prepared from high-speed pellets of 2-day old media from HL23
virus-infected human rhabdomyosarcoma cell cultures. [³H]-DNA
probes for each of the viruses were isolated from the 60-705 RNA:DNA
hybrids of standard large-scale simultaneous detection assays. Hybridization
reactions were set up between the various probes (500-1000 cpm/assay)
and viral RNAs (0.2-1.0 µg) in 20 µl volumes in sealed
siliconized glass tubes in 0.8 M phosphate buffer, pH 6.8,0.1 % SDS
and 10 mM EDT A. After heating at 100 ° C for 1 min, the reactions
were incuba ted
at 68 °C for 20 h (Cot >= 2). The reactions were anlyzed by
thermal elution hydroxyapatite chromatography. Fractions of the total
radioactivity eluted above 60 oC were plotted as a function of temperature.
0 = SSV-1 RNA, black circle = BV-M7 RNA, Q = HL23V RNA.
Table x: Analysis of HL23V for SSV Genomic Content by Competition
Hybridization

Hybridization reactions (5.5 µl) were performed as descibed
in Fig. 9 and contained 0.11 ng 1251-55V RNA (1.2 X 108 cpm/µg),
0.68 ng [³H]-55V cDNA (2 X 10³ cpm/µg) and 0.5-0.25
µg of the indicated competing RNA. Following incubation at
68° for 48 h, the reactions were diluted with 0.01 M Tris-HCI,
pH 8.0,0.4 M NaCI, 0.01 M EDTA and divided into four equal aliquots.
Ribonuclease A (25 units/mI) and ribonuclease T 1 (5 units/ml) were
added to two aliquots and the samples incubated at 37° for 1
h. Nuclease resistance was the ratio of acid-precipitable 125I in
the samples with and without ribonuclease. Recovery of input acid-precipitable
1251 was greater than 90 %. SSV RNA was isolated from virions by
disruption with SDS, Pronase treatment, rate sedimentation in a
sucroseSDS gradient and equilibrium density gradient centrifugation
in potassium iodide. Viral RNAs from GALV-l and MuLV-R were isolated
by similar procedures excepting the KI gradient. HL23V RNA was total
RNA from purified virus and the mouse 18S and 20S RNAs were extracted
from purified ribosomal subunits from N1H/3T3 tissue culture cells.
[³H]-SSV cDNA was synthesized from SSV RNA and oligo dT with
AMV DNA polymerase in the presence of 0.1 mg/µl actinomyc
in D and 0.5 mg/ml distamycin A. The reaction contained TTP, dATP
and dGTP at 1 mM each and [³H]-dCTP (25 Ci/mMole) at 0.05 mM.
This SSV cDNA protected 1251-SSV RNA 33 %, 75 % and 87 % from ribonuclease
digestion at molar input ratios of 0.4,3 and 15, respectively (cDNA:RNA).
Fig. 10: Reconstruction hybridizations. HL23V
[³H]-DNA probe was
hybridized to SSV and BV-M7 RNAs individually and in combination.
0 = SSV RNA, white triangle = BV-M7 RNA,
black circle = SSV and BV-M7 RNAs.
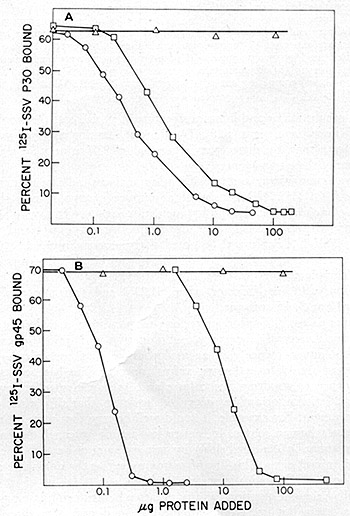
Fig. 11: Competition radioimmunoassays. HL23V was assayed
for the presence of proteins antigenic ally related to the p30 (A)
and gp45 (B) of NC-37 grown SSV-l. Antisera used for the studies with
SSV -p30 were prepared by immunizing rabbits with the purified p30.
An NC-37 absorbed rabbit antiserum prepared by inoculation of disrupted
virions and used for the studies with SSV-gp45 was kindly supplied
by Dr.D.Larson (Pfizer,Maywood, N. J .) .The p30 protein of SSV -1
was purified from sonica ted virus, followed by phosphocellulose and
Sephadex G75 column chromatography and isoelectric focusing. It was
iodinated and repurified and the iodinated p30 antigen preparations
obtained ha.d specific activities of 6 X 10 high 6 cpm/µg. The
gp45 protein of NC-37 grown SSV was purified from sonic-disrupted
virus by column chromatography on agarose 5M in guanidium hydrochloride.
Analysis of the preparations by 5 % sodium dodecyl sulfate polyacrylamide
gel
electrophoresis and isoelectric focusing indicated homogeneity of
the 45,000-dalton antigen. Succinimidyl-3-(4-hydroxphenyl) propionate
(ICN Pharmaceuticals, Inc.) was first labeled with 125I and purified.
The purified gp45 was then labeled by conjugation with the 125I
labeled ester and chromatographed on a Sephadex G-25 column. Titrations
of the antisera were performed in 200 µl reactions that included
0.2 % bovine serum albumin (BSA) in saline, 5000 cpm of 125I-antigen
and dilutions of antibody. After 1 h incubation at 37 °C,
lOO µg of normal rabbit carrier IgG and a titered amount of
goat anti-rabbit IgG were added. The reaction was incubated for 15
hat 4 °C. The samples were centrifuged and both precipitates and
supernatants were counted in a Searle Autogamma counter model 1185.
The results are expressed as percent cpm precipitated. Greater than
80% of the labeled antigen could be bound by specific antisera. Competition
assays were performed in a similar manner, except that unlabeled competing
antigen was added to the original incubation mixture. The unlabeled
competing antigens were SSV = (0), HL23V = (0), SSV-gp45 = (black
circle) and MPMV = (white triangle).
rendered the 125I -SSV completely digestible. The GAL V competed less
successfully, illustrating the sensitivity of this technique for detecting
the small sequence differences known to exist between GAL V and SSV.
HL23V was further tested for its immunological similarity to SSV and
BV-M7. The virus was concentrated by ultracentrifugation from culture
supernatants of A204 (HL23V) and used as competing antigens in radioimmune
assays for the p30 and gp45 of SSV grown in NC-37. As shown in fig.
11, the extent of competition of HL23 V in both of these radioimmune
assa ys was indistinguishable from SSV. Further, an immunodiffusion
analysis of HL23V was made with antisera prepared against the major
and internal structure of proteins of the woolly monkey (p30) and
of the baboon virus (p28). The lines of identity obtained with BV-M7
and SSV indicate that, by these criteria, aga in HL23V cannot be differentiated
from a mixture of these two agents.
In summary, immunologic and hybridization analyses indicate that the
particles produced by A204 (HL23V) consist of a mixture of two viruses
that are indistinguishable immunologically and by nucleotide sequence
from two known nonhuman primate viruses, the baboon endogenous virus
M-7 (61) and the woolly monkey virus SSV-l (62). It will be noted
that our conclusions and results (63) are in complete agreement with
those of Gilden and his collaborators (64) whose experiments, complementary
to ours, involved hybridizations to the cytoplasmic RNA of various
cell lines infected with HL23V and antigenic analysis of the type-specific
p12 and pI5 antigens.
Any attempts to establish productive long-term cultures are always
exposed to the all pervasive danger of laboratory contamination with
animal oncornaviruses. Because of this, any evidence that agents produced
in tissue cultures are either identical or even similar to a known
animal oncornavirus has been accepted as sufficient evidence to condemn
the culture and its particles as irrelevant to the human disease.
I t is important to recognize, however, that this is not a logically
compelling argument. For example, it could well be true that some
animal viruses originated from a human source. It is even less certain
to conclude that an agent is human if it cannot be identified either
by base sequence or by antigenic properties with a known animal virus.
This line of reasoning makes the untenable assumption that our catalogue
of all tumor viruses is complete.
The clinically relevant question for any putative human candidate
particle is not necessarily its origin but its relation to the human
disease. Are there at present any criteria that can be used usefully
to decide whether a given tissue culture virus ( e. g., HL23 V) is
in fact relevant to human neoplasia? A possible resolution can be
achieved by answering the following two questions: (1) Can one provide
evidence at the level of protein and/or nucleotide sequence for the
presence of the putative agent in the original tumor material from
which the tissue culture was established? (2) Can one provide evidence
at the level of protein and/or nucleotide sequence for the persence
of the putative agent in the malignant cells of other patients with
the same disease?
A positive answer to the first question in the form of evidence for
their presence in the original tumor cells would serve to eliminate
the trivial explanation that the particles arose in the culture by
laboratory contamination. The answer to the second question will decide
the general relevance of the observation to human leukemia. Unless
a positive response is obtained in a major portion of the patients
examined, no basis exists for identifying the HL23V particles as clinically
significant agents of human leukemia. We have examined leukemic cells
of 13 patients for the presence of p30 and gp45 of ssv by radioimmune
assays. When these are carried out under conditions that eliminate
non immunologic interference, no evidence for the presence of these
antigens could be detected. This failure would appear to limit the
usefulness of the SSV -p30 as a clinical tool or as a serious etiologic
candidate.
XIII. Purification of Reverse Transcriptase from Human leukemic
Spleens
We have already mentioned the biologic and logistic difficulties
that have attended attempts at obtaining whole human viral particles
for characterization. At the present writing there exists no tissue
culture source of authentic human RNA tumor viruses for use in biochemical
and immunological investigations. One way to obviate these problems
is to forego temporarily the more ambitious goal of characterizing
the whole particle and focus rather on individual protein components
of the human particles. Of these, one of the most accessible is
the reverse transcriptase since its activity can be followed during
fractionation.
The human leukemic reverse transcriptase has been partially purified
from fresh peripheral blood cells obtained from patients with acute
myelogenous leukemia (65, 66). However, the very large amounts of
white blood cells required precluded definitive isolation of the
enzyme in amounts that would establish its purity by gel analysis
and thus permit an unambiguous characterization of its biophysical
and biochemical properties. One way out of this dilemma was to explore
the use of spleens as a source of leukemic enzyme. In addition to
providing a possible solution of the logistic problem, such experiments
would provide useful information on the relation between diseased
tissue and the presence of the virus-like particles. Leukemias often
involve the spleen, and splenectomies are occasionally performed
therapeutically in instances of massive spleen enlargement or persistent
platelet destruction.
We first worked out the technology of enzyme isolation from leukemic
spleens using the murine Rauscher leukemia model. I t was found
that isopycnic separation of virus particles and their conversion
to cores by non-ionic detergents (67, 68) provided material suitably
enriched for enzyme. Usually in such experiments 70 grams of chronic
lymphocytic spleens were used. After thawing, mincing and resuspension,
and homogenization, the extracts were clarified by low speed centrifugation
to remove nuclei and mitochondria. The remaining particulate elements
were then recovered by high speed centrifugation at 80,000 X g for
90 min. Particles were concentrated by isopycnic centrifugation
in sucrose gradients and converted to cores as described previously
(67, 68). After recovery, the cores were disrupted with 1 % NP-40
and 0.7 M KC1 and then subjected to column fractionations on DEAE
cellulose, phosphocellulose, and agarose gels with results as described
in Fig. 12. One main peak of activity is observed on the DEAE column
(Fig. 12A) containing 5 % of the input and greater than 90 % of
the enzyme, yielding a 19-fold enrichment. When the active fractions
of the DEAE cellulose columns are pooled and then chromatographed
on phosphocellulose (Fig. 12B), about 25 % of the protein contains
the enzyme activity, with a recovery of 71 % and an increase in
specific activity of 2.8-fold.
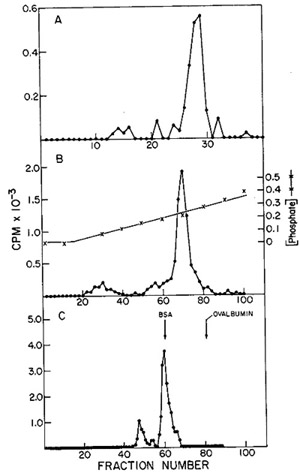
Fig. 12:
(A) DEAE-cellulose chromatography of leukemic spleen enzyme. Core-Iike
particles isolated from a leukemic spleen were treated with 1 %
Nonidet P-40 and 0.7 M KCI, and the solubilized enzyme activity
was chromatographed on a 16.5 cm X 2.5 cm column. Elution was with
0.4 potassium phosphate. 10 µ of each fraction (3.2 ml) were
assayed using on oligo dT-poly rA template.
(B) Phosphocellulose chromatography of leukemic spleen enzyme. The
fractions from the pooled DEAE-cellulose peak activity were diluted
and chromatographed on a 17 cm X 1.5 cm column. Elution was with
160 ml of an 0.01 M-0.05 M potassium phosphate gradient; 1.6.m1
fractions were collected and 10 µI aliquots assayed for oligo
dT-poly rA-templated activity.
(C) Agarose gel filtration of leukemic spleen enzyme. The peak of
activity eluted from phosphocellulose was subjected to gel filtration
on a 50 cm X 0.9 cm agarose column. The elution rate was 4 ml/hr
and 0.4 ml fractions were collected. Aliquots of 4 µI were
assayed for enzyme activity with an oligo dT-poly rA template.
The concentrated phosphocellulose enzyme is then passed through
a 0.5 M agarose column on which one routinely observes two peaks
of activity (fig. 12C). If the first peak of activity is rechromatographed
on the same type of column, a shift of most of the activity to the
position of the second peak occurs. Thus, the first peak would appear
to be an aggregate (possibly a dimer) of the enzyme. A summary of
the column fractionations in terms of recoveries and specific activities
is recorded in Table XI.
Table XI: Purification of RNA-dependent DNA Polymerase from
a Viral Core Fraction of Human Leukemic Spleen
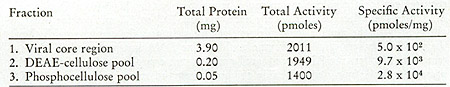
Fig. 13: Sodium dodecyl sulfate-polyacrylamide gel agarose
enzymes. Fractions 48(A) and 60(B) of Fig. 12C were subjected to
electrophoresis on sodium dodecyl sulfate polyacrylamide gels. After
staining with Coomassie blue, the gels were scanned in a Gilford
Model 2400 spectrophotometer. The stained gel of each fraction is
also shown.
To examine the purity of the final enzyme preparation, portions of
the two agarose peak fractions of Fig. 12C were electrophoresed on
5 % acrylamide gels in the presence of 0.1 0/0 sodium dodecyl sulfate.
Molecular weights of the
separated polypeptides were determined using lysozyme, ovalbumin,
bovine serum albumin and aldolase as molecular weight markers. As
shown by the gel photographs and the scans of figures 12A and 12B,
both agarose peaks yielded one major band corresponding to a molecular
weight of 70,000 daltons, supporting the conclusion that the first
agarose peak was in fact an aggregate of the second. A pool of the
two agarose peaks constituted the final enzyme preparation.
Several parameters of the reverse transcriptase were examined. The
reaction was found to proceed best at 37° and over abroad pH range
from 7.0 to 8.2. The requirement for a divalent cation could be satisfied
best by Mg++ (6 mM) although Mn++ was also effective over a very narrow
range (0.5 to 1.2 mM), a feature also observed with the murine leukemic
reverse transcriptases. In common with these, the purified leukemic
spleen enzyme could utilize DNA but preferred RNA as a template. Further,
the RNA templated activity was completely dependent on the presence
of all four deoxyriboside triphosphates and exogenously added RNA
(Table XII).
Table XII: Deoxynucleoside Triphosphate and RNA Requirements
of Leukemic RNA-dependent DNA Polymerase

The critical diagnostic criterion of a putative reverse transcriptase
is the ability to transcribe heteropolymeric RNA into a DNA complement.
To test this, a reaction was run with the purified enzyme employing
isolated AMV-RNA as the template. The product synthesized was purified,
alkali-treated and hybridized either to AMV or RL V-RNA. An analysis
on cesium sulfate gradient showed clearly that the tritiated product
was complementary only to AMV-RNA. Thus, the enzyme purified from
the human leukemic particles satisfied this operational definition
of a reverse transcriptase.
The results (69) we have just described represent the first instance
of a human reverse transcriptase isolated to the purity required
for complete characterization. In addition, the procedure provides
enough protein to generate monospecific antisera for ultimate use
as detecting devices.
XIV. Present Status and Future Prospects
The application of nucleic acid hybridization and the other techniques
of molecular biology have permitted us to illuminate some of the
issues of human cancer noted in the questions listed in the introductory
paragraphs. The answers obtained may be summarized in the following
statements:
(1) Human neoplasias do contain RNA molecules possessing detectable
homologies to the RN A of tumor viruses known to cause similar cancers
in animal systems .
(2) The RNA molecules indentified in the human tumors possess the
size and physical association with reverse transcriptase that characterizes
the RNA of the animal oncornaviruses .
(3) Further, the tumor-specific RNA is encapsulated in a particle
possessing the size and density of the RNA tumor viruses .
(4) The RNA of the human tumor particles possess homology to the
RNA of the viruses causing the corresponding diseases in animals
.
(5) Viral-related sequences are unique to the DNA of tumor cells,
are inserted postzygoticall y, and are therefore not resident in
the germ line. The availability of the mesenchymal and mammary tumor
animal model systems provided us with the viral agents that generated
the radioactive DNA probes used to search for and find the homologous
sequences in the particulate fractions of the corresponding human
neoplasias. Our invention and perfection of the "simultaneous detection
test" enabled us to characterize the size and particulate nature
of the RNA detected in the human tumors. Beyond this, it evolved
into an important technical advance since it permitted us to bypass
the restriction of being forced to start with a known animal viral
agent. Thus, we were able to extend our investigations beyond breast
cancer and the mesenchymal tumors to other clinically important
human neoplasias. The use of the simultaneous detection test established
that a variety of human carcinomas contained particles having the
diagnostic criteria associated with the oncornavirus-like particles.
The neoplasias examined included a variety of cancers of brain (70),
the gastrointestinal tract and lung (71 ), and skin (72).
Having found these particles in the human tumors, where does one
go from there? An obvious need is to characterize them more fully
biochemically and immunologically so that they can be compared with
the similar agents found in the animal systems. However, here we
are faced with a logistical problem that has thus far interposed
serious obstacles. The amount of tumor material available and its
particle content are such that it is difficult to isolate particles
in sufficient quantity and purity to perform the desired biochemical
examinations. Adequate , characterizations will not be readily feasible
until these particles are obtainable in suitable yield from established
cell lines. To date this desideratum has not been achieved. Nevertheless,
it may still prove possible to exploit some of the implications
derivable from certain of their features. In particular, at least
a portion of I the RNA sequences found in the particles associated
with each primary tumor site ,1 appears to be unique. As we have
already noted, the particles found in human " breast cancer share
no sequences in common with those found in the mesenchymal tumors.
These differences appear to extend to the particles found in other
sorts or primary tumors. Thus, we have been able to distinguish
by cross hybridization the sequences found in particles from stomach
cancers from those found in the colon. Similarly, the sequences
found in lung cancer particles were easily differentiated from those
found in other tumor sites tested. It would appear from our survey
that the sequences found in these particles are histogenic ally
specific.
These results suggest the possibility of developing a novel pathway
for specific tumor detection. From the outset, it was evident that
the nucleic acid hybridization technology we had developed and used
to provide fundamental information of the molecular basis of the
cancer cell was not likely to be useful in a clinical setting. In
the first place, it has thus far been successfully applied only
to tumor cells, and there is little likelihood that there would
be enough circulating tumor-specific nucleic acids to be useful
for diagnostic purposes. Further, the hybridization procedure is
too sophisticated, too laborious, and too expensive to be introduced
into the clinical pathology laboratory.
It seems clear that one must find a way of translating the sequence
differences of the tumor particles into a parameter that would be
more amenable to detection by the devices more commonly used in
the clinical laboratory. One approach would depend upon the plausible
expectation that the sequence differences observed in the different
particles would be reflected in proteins that might be distinguishable
antigenically. Were this realized, one could immediately hope to
use the very sensitive and less restrictive methods of immunology.
These are not only very sensitive, but they are in routine use in
clinical laboratories.
To exploit this approach and obviate the logistic and other difficulties
attending attempts to study the whole particles, one might focus
rather on isolating and characterizing individual protein components.
Of these, one of the most amenable is the reverse transcriptase
since it can be followed during fractionation by means of its enzyme
activity. As we have shown here, this approach has led to the successful
isolation and purification of the reverse transcriptase of the particles
found in leukemic spleens. These same methodologies can be and have
been applied to other human neoplastic tissue. With these specific
proteins available, monospecific antisera can be generated that
could hopefully be used for detection in the body fluids of tumor-bearing
individuals. The potential value of providing a use able specific
assay for the presence of tumor cells has been even further enhanced
by the recent advances in adjuvant chemotherapy. Efforts along these
lines could convert what has thus far been an exercise in molecular
biology into a potentially powerful clinical tool.
Acknowledgments
Supported by grant CA-02332 from the U.S. Public Health Service
and by
Virus Cancer Program contract NO1-6-1010 with the National Cancer
Institute.
References:
1. Hall, B. D., and Spiegelman, S. (1961) Proc. Nat. Acad. Sci.
USA 47, 137-146.
2. Axel, R., Schlom, J. and Spiegelman, S. (1972) Nature 235,32-36.
3. Axel, R., Schlom, J. and Spiegelman, S. (1972) Proc. Nat. Acad.
Sci. USA 69, 535-538.
4. Schlom, J., Spiegelman, S. and Moore, D. H. (1971) Nature 231,97-100.
5. Hehlmann, R., Kufe, D. and Spiegelman, S. (1972) Proc. Nat. Acad.
Sci.USA 69, 435-439.
6. Kufe, D., Hehlmann, R., and Spiegelman, S. (1972) Science 175,
182-185.
7. Hehlmann, R., Kufe, D. and Spiegelman, S. (1972) Proc. Nat. Acad.
Sci. USA 69, 1727-1731.
8. Gallo, R. C., Miller, N. R., Saxinger, W. C. and Gillespie, D.
(1973) Proc. Nat.Acad. Sci. USA 70, 3219-3224.
9. Vaidya, A. B., Black, M. M., Dion, A. S. and Moore, D. H. (1974)
Nature 249, 565-567.
10. Baltimore, D. (1970) Nature 226, 1209-1211.
11. Temin, H. M., and Mizutani, S. (1970) Nature 226, 1211-1213.
12. Spiegelman, S., Hall, B. D. and Storck, R. (1961) Proc. Nat.
Acad. Sci. USA 47, 1135-1141.
13. Rokutanda, M., Rokutanda, H., Green, M., Fujinaga, K., Ray,
R. K. and Gurgo, C. (1970) Nature 227, 1026-1029.
14. Spiegelman, S., Burny, A., Das, M. R., Keydar, J., Schlom, J.,
Travnicek, M.and Watson, K. (1970) Nature 227, 563-567.
15. Schlom, J., and Spiegelman, S. (1971) Science 174, 840-843.
16. Schlom, J., Spiegelman, S. and Moore, D. H. (1972) Science 175,
542-544.
17. Gulati, S. C., Axel, R. and Spiegelman, S. (1972) Proc. Nat.
Acad. Sci. USA 69, 2020-2024.
18. Axel, R., Gulati, S. C. and Spiegelman, S. (1972) Proc. Nat.
Acad. Sci. USA 69, 3133-3137.
19. Baxt, W., Hehlmann, R. and Spiegelman, S. (1972) Nature New
BioI. 240, 72-75.
20. Spiegelman, S., Kufe, D., Hehlmann, R. and Peters, W. P. (1973)
Cancer Research 33, 1515-1526.
21. Kufe, D., Margrath, I. T., Ziegler, J. L. and Spiegelman, S.
(1973) Proc. Nat. Acad. Sci. USA 70, 737-741.
22. Todaro, G. J. and Huebner, R. J. (1972) Proc. Nat. Acad. Sci.
USA 69, 1009-1015.
23. Ruprecht, R. M., Goodman, N. C. and Spiegelman, S. (1973) Proc.
Nat. Acad. Sci. USA 70, 1437-1441.
24, Baxt, W. G. and Spiegelman, S. (1972) Proc. Nat. Acad. Sci.
USA 69, 3737-3741.
25. Kufe, D. W., Peters, W. P. and Spiegelman, S. (1973) Proc. Nat.
Acad. Sci. USA 70, 3810-3814.
26. Kufe, D., Hehlmann, R. and Spiegelman, S. (1973) Proc. Nat.
Acad. Sci. USA 70, 5-9.
27. Henle, G., Henle, W., Clifford, P., Diehl, V., Kafuko, G., Kirya,
B., Klein, G.Morrow, R., Munube, G., Pike, P., Tukel, P. and Ziegler,
J. (1969) J. Nat. Cancer Inst. 43, 1147-1157.
28. zur Hausen, H., Schulte-Holthausen, H., Klein, G., Henle, W.,
Henle, G., Clifford, P. and Santesson, L. (1970) Nature 228, 1056-1058.
29. Henle, G., Henle, W. and Diehl, v. (1967) Proc. Nat. Acad. Sci.
USA 59) 94-101.
30. Niederman, J. C., Evans, A. S., Subrahmanyan, L. and McCollum,
R. W. (1970) New Eng. J. Med. 282, 361-365.
31. Geh, K. and Swisher, S. N. (1965) Arch. Int. Med. 115, 475-478.
32. Geh, K., Swisher, S. N. and Herman, E. C., Jr. (1967) Arch.
Int. Med. 120) 214-219.
33. Baxt, W., Yates, J. W., Wallace, H. J., Jr., Holland, J. F.
and Spiegelman, s. ( 1973) Proc. N at. Acad. Sci. USA 70, 2629-2632.
34. Goodman, N. C., Ruprecht, R. M., Sweet, R. W., Massey, R., Deinhardt,
F. and Spiegelman, S. (1973) Int. Jour. Cancer 12, 752-760.
35. Sweet, R. W., Goodman, N. C., Cho, J.-R., Ruprecht, R. M., Redfield,
R., R. and Spiegelman, S. (1974) Proc. Nat. Acad. Sci. USA 71, 1705-1709.
36. Chattopadhyay, S. K., Rowe, W. P., Teich, N. M. and Lowy, D.
R. (1975) Proc. Nat. Acad. Sci. USA 72, 906-910.
37. Jaenisch, R., Fan, H. and Croker, B. (1975) Proc. Nat. Acad.
Sci. USA 72, 4008-4012.
38. Frei, E. and Freireich, E. J. (1965) Adv. in Chemotherapy 2,
269-298.
39. Simone, J., Aur, R. J." Hustu, H. 0. et al. (1972) Cancer
30, 1488-1494.
40. Reisman, L. E., Zuelzer, W. W. and Thompson, R. I. (1964) Cancer
Res. 1448-1460.
41. Sandberg, A. A., Takaaki, J., Kikuchi, Y. et al. (1964) Ann.
N. Y. Acad. Sci. 113, 663-716.
42. Whang-Peng, J., Freireich, E. J., Oppenheim, J. J. et al. (1969)
J. Nat. Cancer Inst. 42, 881-897.
43. Trujillo, J. M., Cork, A., Hart, J. S. et al. (1974) Cancer
33, 824-833.
44. Duttera, M. J., Whang-Peng, J., Bull, J. M. C. et al. (1972)
Lancet 1,715-717.
45. Craddock, C. G. and Crandall, B. F. (1973) Blood 42,1013.
46. Greenberg, P. L., Nichols, W. C. and Schrier, S. L. (1971) New
Eng. J. Med. 284, 1225-1232.
47. Harris, J. and Freireich, E. J. (1970) Blood 35,61-63.
48. Moore, M. A. S., Williams, N. and Metcalf, D. (1973) J. Nat.
Cancer Inst. 50) 603-623.
49. Bull, J. M., Duttera, M. J., Stashick, E. D. et al. (1973) Blood
42,679-676.
50. Gutterman, J. U., Mavligit, G., Burgess, M. A. et al. (1974)
J. Nat. Cancer Inst. 53,389-392.
51. Halterman, R. H., Leventhal, B. and Mann, D. L. (1972) New Eng.
J. Med. 287, 1272-1274.
52. Killmann, S. A. (1968) Ser Haemat 1,103-128.
53. Mak, T. W., Aye, M. T., Messner, H. et al. (1974) Brit. J. Cancer
29,433-437.
54. Mak, T. W., Manaster, J., Howotson, A. F. et al. (1974) Proc.
Nat. Acad. Sci. USA 71, 4336-4340.
55. Fischinger, P. J., Nomura, S., Peebles, P. T. et al. (1972)
Science 176, 1033-1035.
56. McGrath, C. M., Grant, P. M., Soule, H. D., Glancy, T. and Rich,
M. A. (1974) Nature 252, 247-250.
57. Kotler, M., Weinberg, E., Haspel, 0., Olshevshy, U. and Becker,
Y. (1973) Nature New BioI. 244, 197.
58. Gallagher, R. E. and Gallo, R. C. (1975) Science 187, 350-353.
59. Teich, N. M., Weiss, R. A., Salahuddin, S. Z., Gallagher, R.
E., Gillespie, D. H. and Gallo, R. C. (1975) Nature 256, 551-555.
60. Gallo, R. C., Gallagher, R. E., Miller, N. R., Mondal, H., Saxinger,
W. C., Mayer, R. J., Smith, R. G. and Gillespie, D. H. (1974) Cold
Spring Harbor Symposium on Quantitative Biology XXXIX, 933-961.
61. Benveniste, R. E., Lieber, M. M., Livingston, D. M., Sherr,
C. J. and Todaro, G. J. (1974) Nature 248, 17-20.
62. Theilen, G. H., Gould, D., Fowler, M. and Dungworth, D. L. (1971)
J. Nat. Cancer Inst. 47, 881-889.
63. Chan, E., Peters, W. P., Sweet, R. W., Ohno, T., Kufe, D. W.,
Spiegelman, S., Gallo, R. C. and Gallagher, R. E. (1976) Nature
260, 266-268.
64. Okabe, H., Gilden, R. V., Hatanaka, M., Stephenson, J. R., Gallagher,
R. E., Gallo, R. C., Tronick, S. R. and Aaronson, S. A. (1976) Nature
260, 264-266.
65. Sarngadharan, M. G., Sarin, P. S., Reitz, M. S. and Gallo, R.
C. (1972) Nature New BioI. 240, 67-72.
66. Mondal, H., Gallagher, R. E. and Gallo, R. C. (1975) Proc. Nat.
Acad. Sci. USA 72, 1194-1198.
67. Feldman, S. P., Schlom, J. and Spiegelman, S. (1973) Proc. Nat.
Acad. Sci. USA 70, 1976-1980.
68. Michalides, R., Spiegelman, S. and Schlom, J. (1975) Cancer
Res. 35, 1003-1008.
69. Witkin, S., Ohno, T. and Spiegelman, S. (1975) Proc. Nat. Acad.
Sci. USA 72, 4133-4136.
70. Cuatico, W., Cho, J.-R. and Spiegelman, S. (1973) Proc. Nat.
Acad. Sci. USA 70, 2789-2793.
71. Cuatico, W., Cho, J.-R. and Spiegelman, S. (1974) Proc. Nat.
Acad. Sci. USA 71, 3304-3308.
72. Balda, B.-R., Hehlmann, R., Cho, J.-R. and Spiegelman, S. (1975)
Proc. Nat. Acad. Sci. USA 72,3697-3700.
|